4 Making a Phylogenetic Tree
4.1 Introduction
In this section, you will start with a set of bacterial protein sequences and use these to construct and visualise a phylogenetic tree, using online bioinformatics software tools. To do this you will move through the following stages:
- Acquire sequence data
- Align sequences
- Construct a tree from the alignment
- Visualise the tree
Although the principles and the order of actions taken here are the same as we would use at a research level, most research-level phylogenetic reconstruction you will see is likely to use a different set of tools than those employed here.
4.1.1 Scenario
You are a biomedical researcher interested in evolution of the pathogen Burkholderia pseudomallei, which causes melioidosis in humans (Chewapreecha et al. (2017)).
Depending on whether you live in a medically well-resourced, or poorly-resourced, area the risk of death from infection varies from 10%-40%. The pathogen itself has the capability to infect a range of human cells and evade the immune system, in part because it produces proteins called effectors: these are proteins adapted to interfere with host biology and make the environment more favourable for the bacterium.
The protein you are studying, BipC, is such an effector protein. Specifically, it is a translocator protein that makes holes in the host’s cells, allowing its own effector proteins to enter and manipulate host biochemistry, and also binds to actin microfilaments, disrupting host cell structure.
You would like to understand the evolution of this protein, to see whether the knowledge can help you design drug compounds that interfere with the action of BipC as a treatment for melioidosis. To do this, you will produce a phylogenetic tree for the protein.
4.2 Obtain sequences from UniProt
The UniProt database is the most comprehensive protein sequence database, combining protein sequence, structure, and annotation data. UniProt has an entry for a relative of the protein you are interested in, which has accession Q62B08.
Go to the UniProt database link and find the record associated with accession number Q62B08.
On the main UniProt landing page, enter the accession of the sequence you are searching for in the field under Find your protein and click Search
You should find yourself at the page for Q62B08, BIPC_BURMA.

Q62B08 page header.
4.2.1 Exploring the UniProt record
The UniProt database can provide a great deal of information about the proteins it contains. This can be a great help in understanding more about your protein, and finding useful links to other sources of information. To help you get greater knowledge about BipC, please answer the formative questions below in the MyPlace quiz
What organism does this sequence come from?
Look at the header for the record page.
How many amino acids does this protein contain (i.e. how long is the sequence)?
Look at the header for the record page, or click on Sequence in the sidebar.
According to the Gene Ontology (GO), what is the subcellular location of this protein?
Click on Subcellular Location in the sidebar, and then look at the GO Annotation tab.
How many identical (100% identity) sequences are there in the UniProt database?
Click on Similar Proteins and look at the table.
What are the accessions of the identical sequences in Burkholderia pseudomallei
Can you find this organism in the Similar Proteins table?
4.2.2 Downloading sequence homologues
We only construct phylogenetic trees from protein sequences that are related to each other - it wouldn’t make much sense to try to infer relationships between unrelated sequences!. When we say that two sequences are related we mean that they share a recognisable common ancestor.
Two sequences that share a common ancestor are said to be homologous.
Homology - the state of two sequences sharing a common ancestor - is an all-or-nothing state. Two sequences are either homologous to each other, or they are not. It is incorrect to talk about gradation or percentage of homology (though you will encounter people doing so).
UniProt provides a precomputed cluster of all the proteins it knows about that are potentially homologous to any other protein sequence in its database, and you can find this by clicking on the Similar Proteins link in the sidebar of the record page. This will take you to a table of similar protein sequences (Figure 4.4).
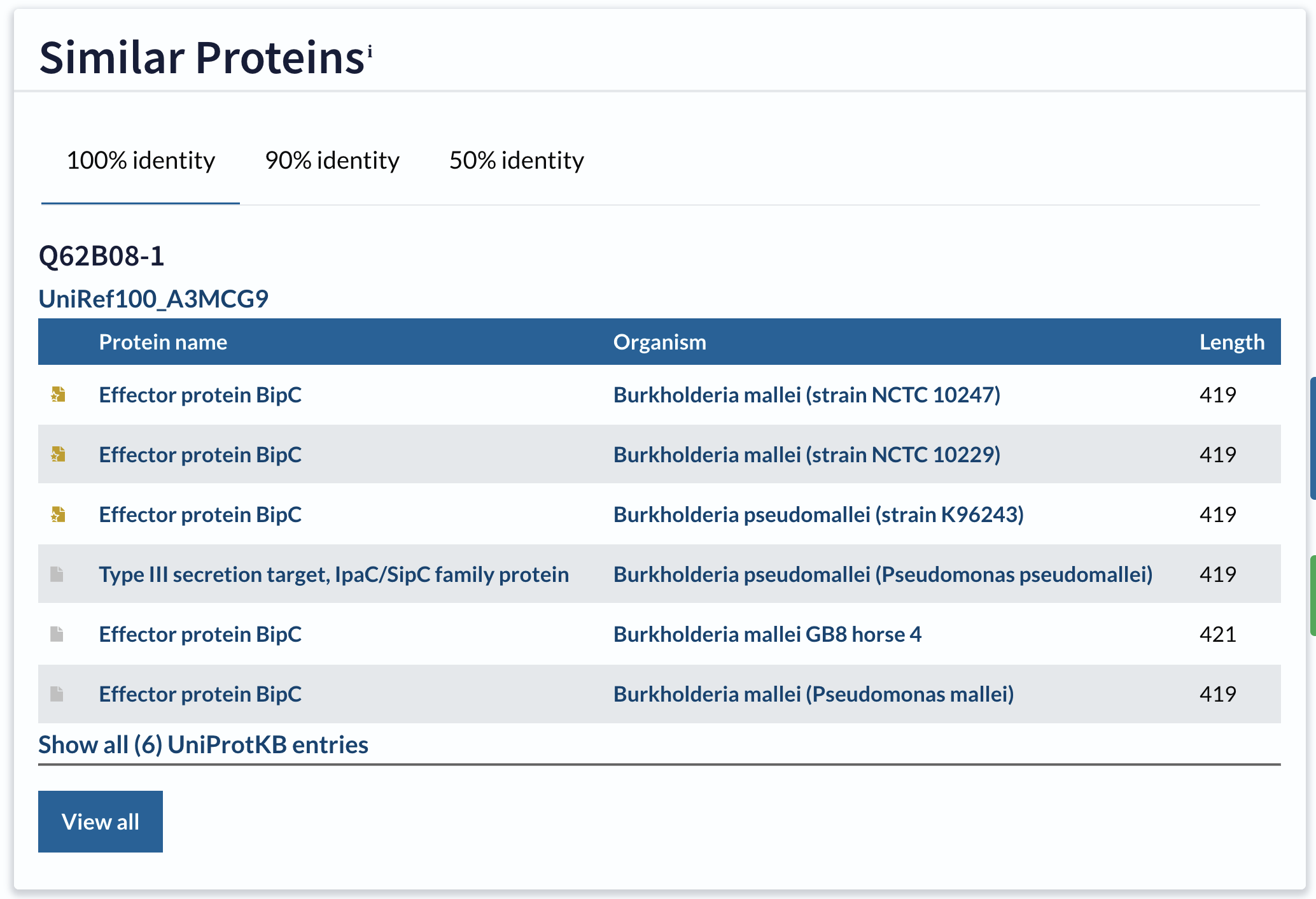
Similar Proteins table for Q62B08
This table has three tabs: 100% identity, 90% identity, and 50% identity. Clicking on these will show you the clusters of sequences in UniProt sharing at least that level of sequence identity with the current record. In general, the number of sequences in the cluster increases as the percentage identity used to gather the cluster goes down.
View all sequences in the 50% identity cluster for Q62B08.
- On the UniProt record page for
Q62B08click on theSimilar Proteinslink in the sidebar to get the similar proteins table. - Click on the
50% identitytab to see the first few proteins in the cluster. - Click on
View Allto see the full list of sequences in the cluster.
You should find yourself at the UniRef cluster page with 50% identity to Q62B08.
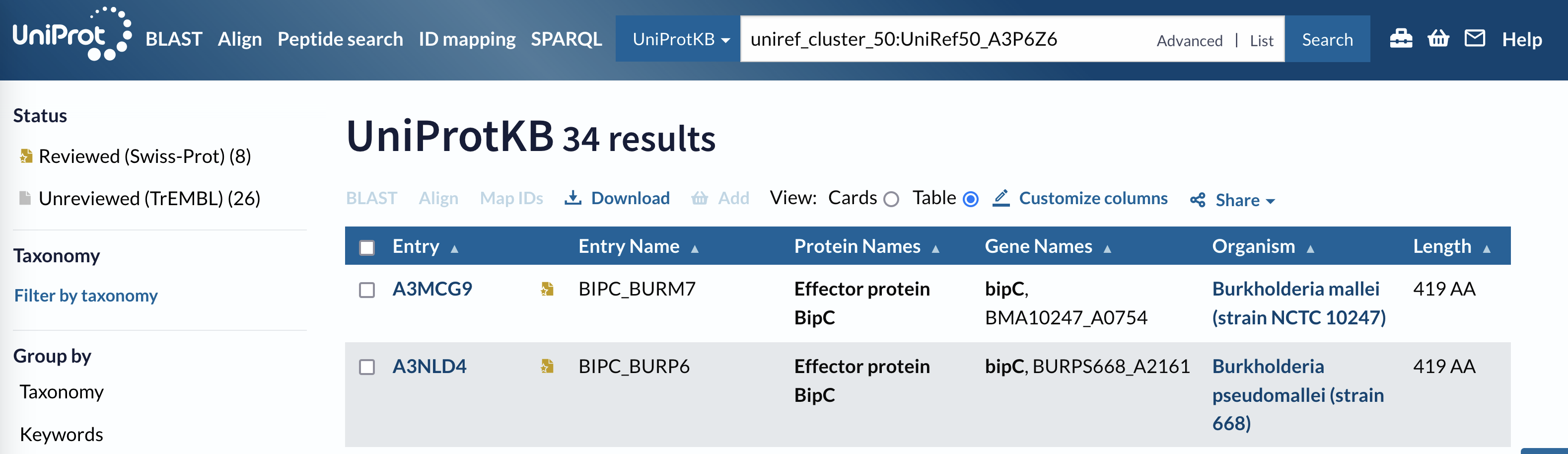
Q62B08 50% sequence identity cluster page header.
This set of sequences are all closely-enough related to each other for drawing a phylogenetic tree to be worthwhile. Several different species are represented, which should make it an informative tree in regards to the evolution of this protein effector. You will use this set of sequences to construct your tree.
Download all sequences (not compressed) in the 50% identity cluster for Q62B08 to your computer.
- Click on the
Downloadlink at the top of the table. This will bring up a set of options (Figure 4.6). - Make sure
Download allandCompressed - Noare selected. - Click on
Downloadand save the file somewhere you can find it again (e.g. on yourDesktop).
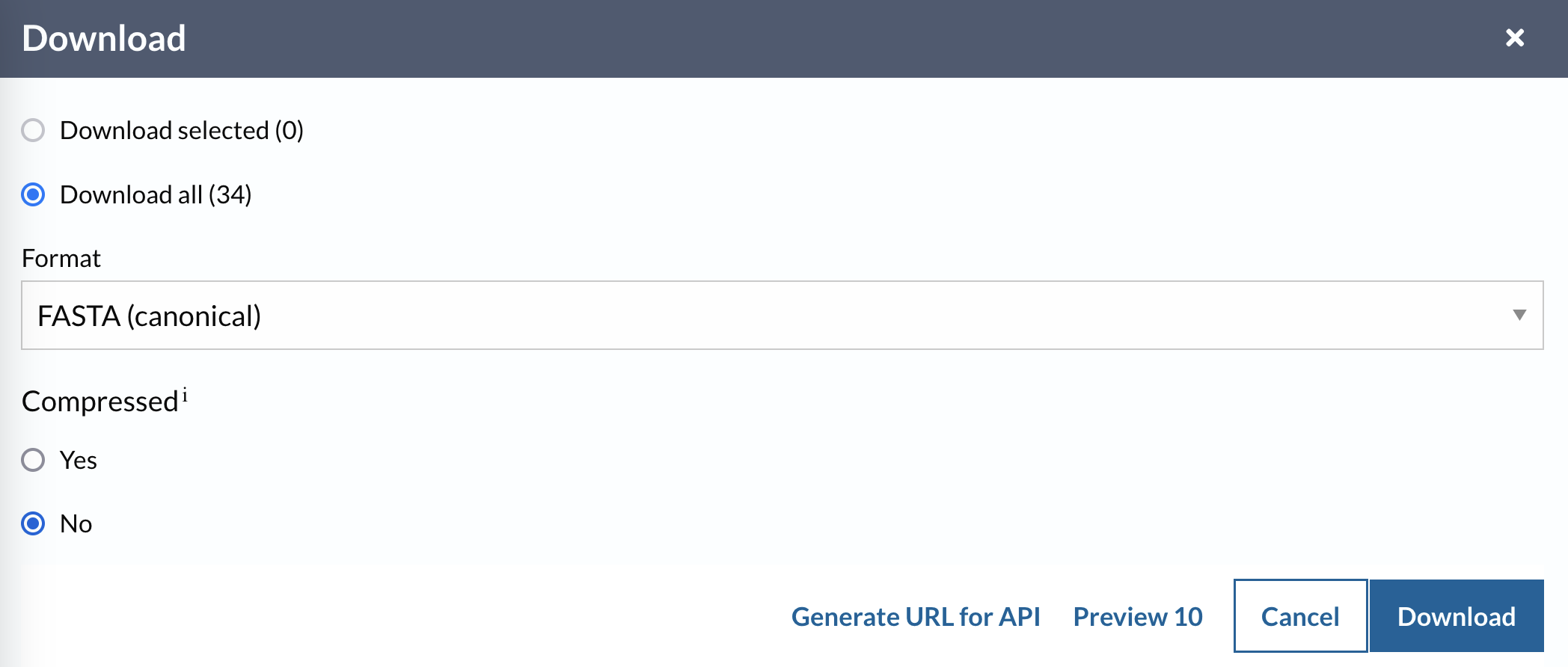
Your sequence file should be the same as that which you can download from this link.
Please answer the formative questions below in the MyPlace quiz
How many sequences are there in this cluster?
Look at the header for the cluster page.
How long is the shortest sequence in this cluster?
Look in the Length column of the cluster table.
How many species are represented in the dataset?
Look in the Organism column of the cluster table.
4.3 Make a multiple sequence alignment
The sequences in the cluster you have downloaded from UniProt are different lengths, and are not identical to each other.
If we were to compare animal body parts to understand their evolution, we would naturally try to compare only equivalent elements, such as forelimbs, with each other. It wouldn’t be reasonable to compare organ structure with skull shape, say.In the same way, for biological sequence data, we want to compare genetic changes at equivalent sites, which means we have to find which sites are equivalent before we build the tree.
We find equivalent sites by aligning sequences against each other in a Multiple Sequence Alignment (MSA). Detailed accounts of the methods involved in multiple sequence alignment are beyond the scope of the course, but they can be similar in some cases to the pairwise sequence alignment process that you have already seen in BM214.
You are going to employ a widely-used research tool called MUSCLE to make a multiple sequence alignment from the sequences in the cluster you downloaded. This program is availble to install on your own computer, but can also be used through an online service (Figure 4.7).
MUSCLEonline service: https://www.ebi.ac.uk/jdispatcher/msa/muscle
MUSCLE online service landing page.
Use the online MUSCLE service to generate a multiple sequence alignment from your cluster sequences.
- On the
MUSCLEservice landing page, click onBrowse…and use the dialogue box that appears to navigate to your downloaded cluster sequences. - Scroll down the page and click
Submit
Your alignment should look similar to the one in Figure 4.8.
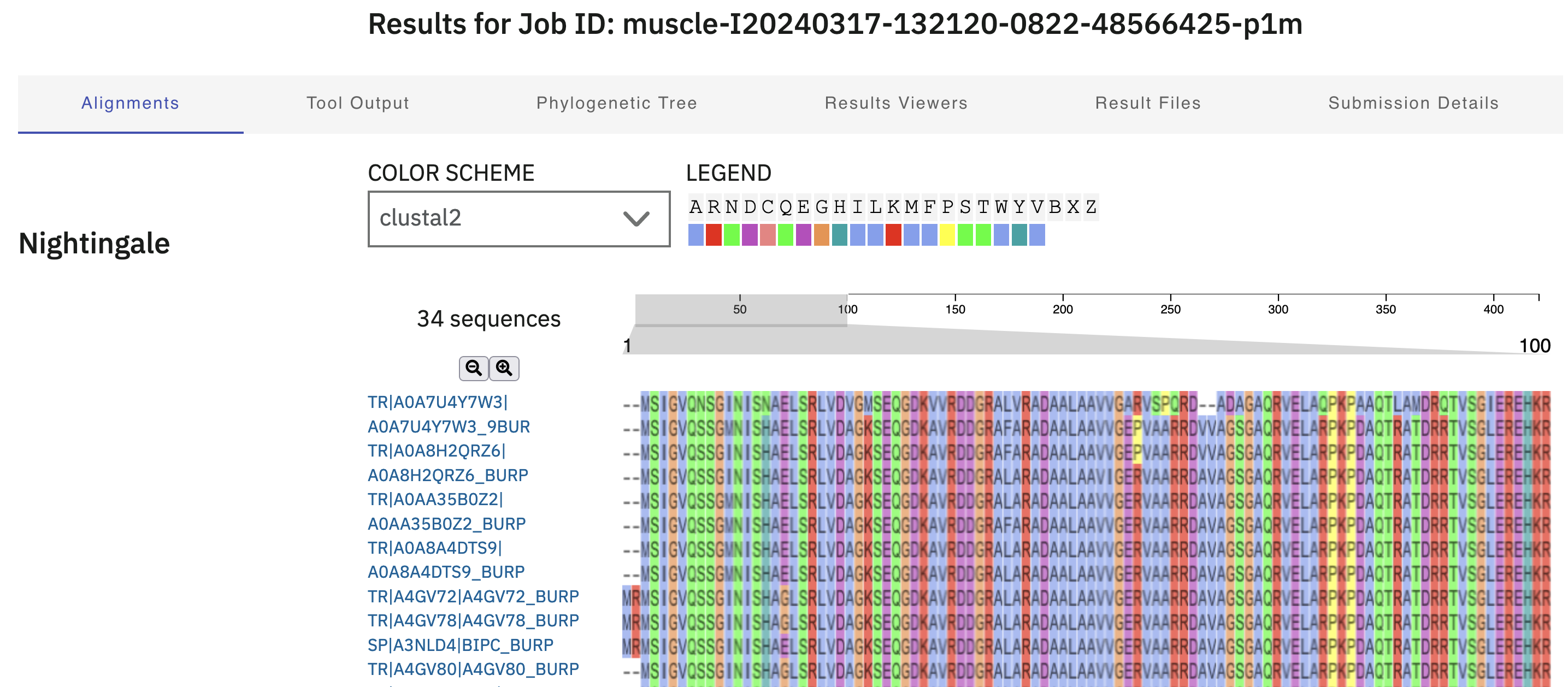
MUSCLE multiple sequence alignment.
The output from the online tool can be very helpful. You can use your mouse to scroll around the alignment visualisation to see which regions are similar, and which differ. There are also options for colouring the residues in the alignment by a range of schemes, to highlight potentially conserved biochemical properties of the residues (Figure 4.9).
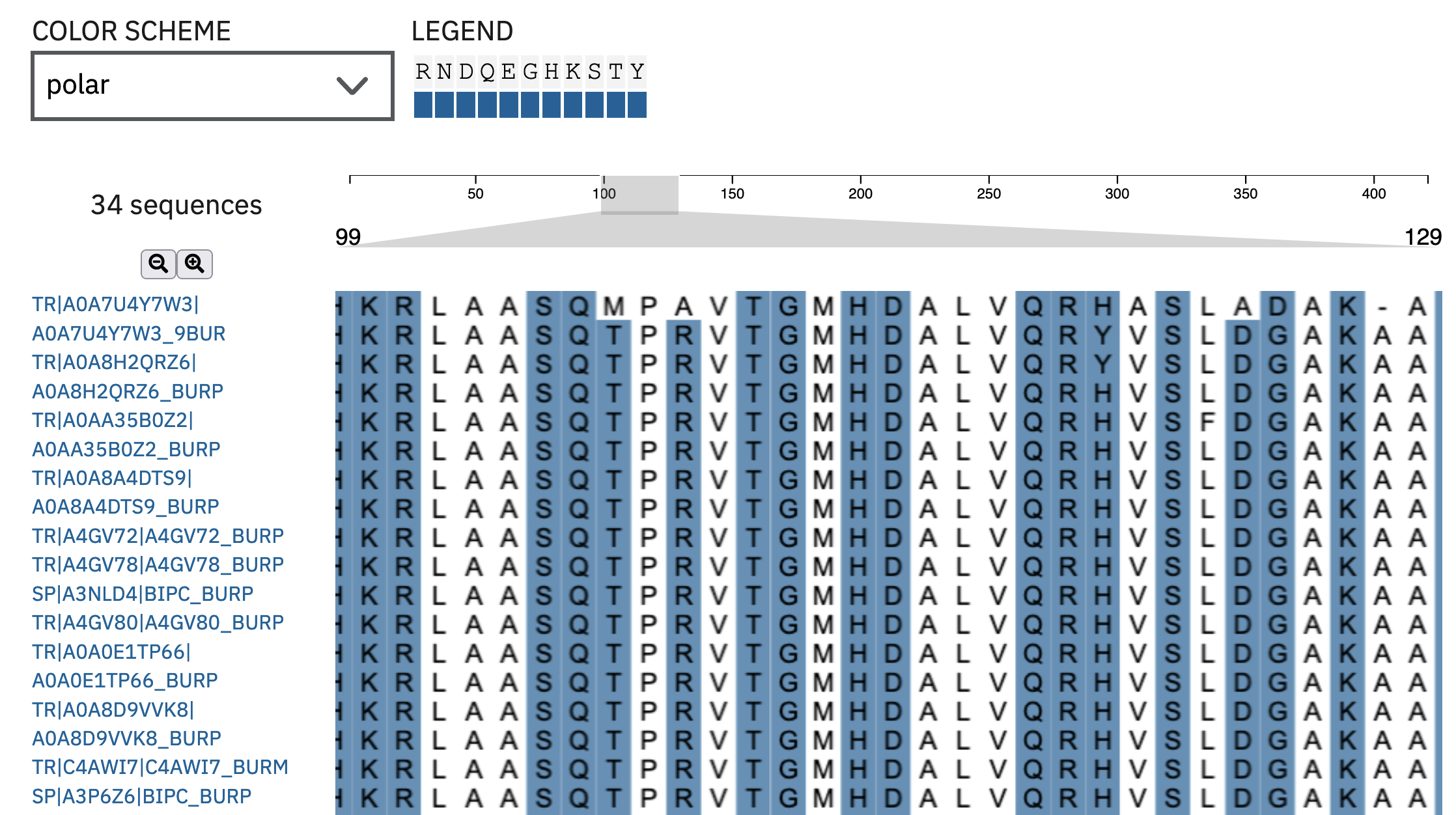
MUSCLE multiple sequence alignment highlighting polar residues.
Download the CLUSTAL format MUSCLE sequence alignment file to your computer.
- On the sequence alignment page, click on the
Result Filestab. - Find the
Alignment in CLUSTAL formatline in the table, click on the `Download button next to it, and use the dialogue box to save the file somewhere you can find it.
Your alignment file should be the same as the one you can download from this link
4.4 Construct a phylogenetic tree
Now you have a sequence alignment, you are ready to build a phylogenetic tree.
For this workshop we are using a relatively simple tool that will give us a phylogenetic tree quickly. The approach we are using here is known to have systematic issues and would not be used in a modern research setting. However, it is a method that was very widely-used until the early 2000s and is still sometimes seen in current research.
You are going to use a tool called Simple Phylogeny to make a phylogenetic tree from your multiple sequence alignment (Figure 4.10). This tool implements tree generation methods from the ClustalW software tool, and is restricted to two relatively basic clustering approaches:
We will use this program via its online service (Figure 4.10)
Simple Phylogenyonline service: https://www.ebi.ac.uk/jdispatcher/phylogeny/simple_phylogeny)
Simple Phylogeny online service landing page.
Use the online Simple Phylogeny service to generate a Neighbour-Joining phylogenetic tree from your alignment.
- Open the sequence alignment file you downloaded in a text editor (Use Notepad or similar - DO NOT USE MICROSOFT WORD).
- Copy the data from the file and paste it into the
Input Sequencefield. - Set Tree Format to
Clustal - You can leave all the other options as their default settings (Figure 4.11).
- Scroll down the page and click
Submit
Simple Phylogeny tree generation.
The output should look similar to the tree data in Figure 4.12. Note that this file is in .newick format, but puts each parenthesis (() on a new line.
Simple Phylogeny tree output.
Download the .newick format tree file to your computer.
- On the
Simple Phylogenyoutput page, click on theResult Filestab. - Find the
Phylogenetic Treeline in the table, click on the `Download button next to it, and use the dialogue box to save the file somewhere you can find it.
Your tree file should be the same as that which you can download from this link
4.5 Visualise the tree
You will now use iTol to visualise the tree you produced. Use what you have learned from Chapter 2 to view the tree.
Use iToL to visualise the .newick tree file you generated with Simple Phylogeny.
- Click on the
Uploadbutton iniTol - Click on the
Browse…button and use the dialogue box to navigate to the location of the tree file you downloaded. - Click the
Uploadbutton
The tree file shown in iTol should resemble Figure 4.13.
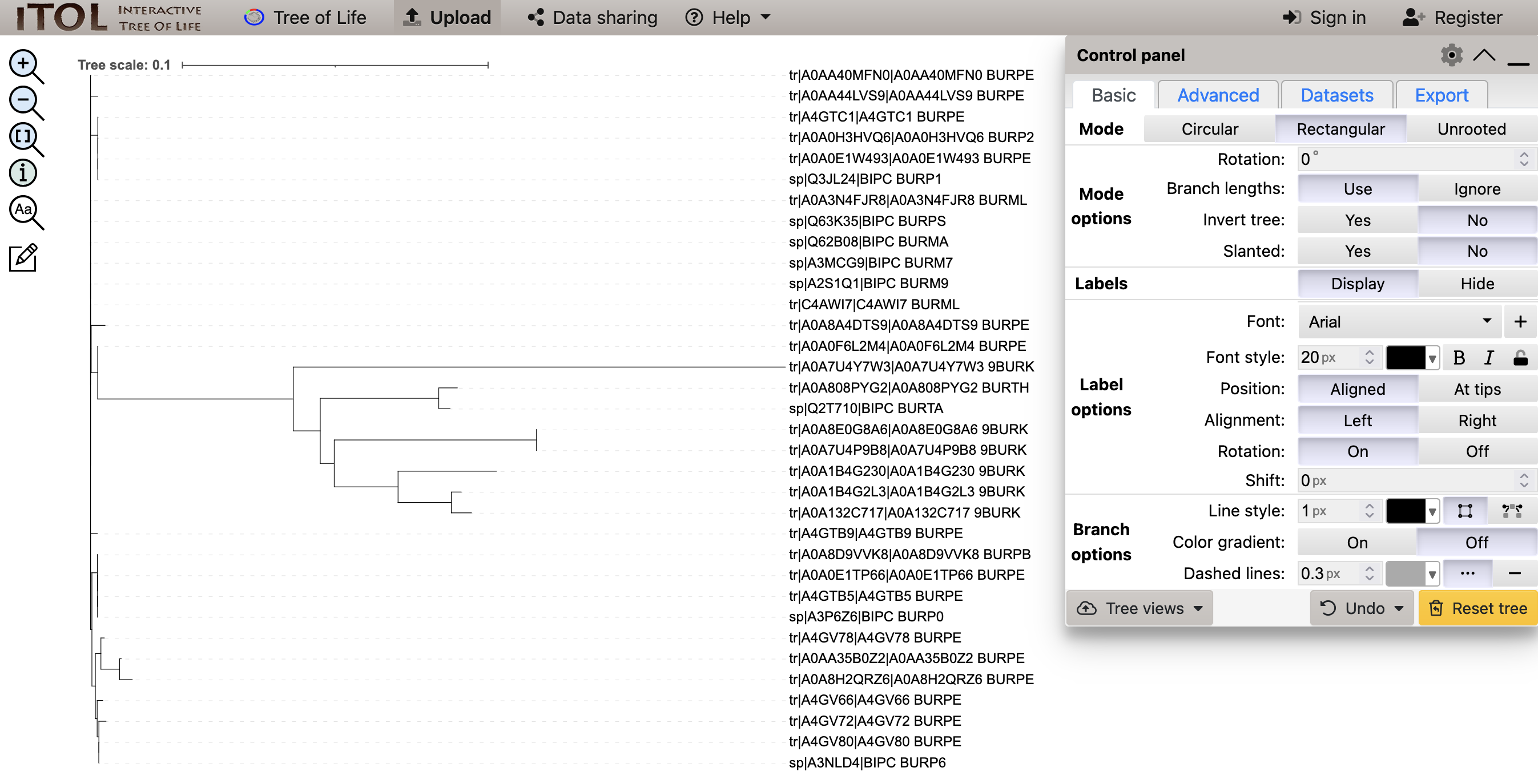
Simple Phylogeny, visualised in iTol
4.6 Summary
After successfully working through this section you should be able to:
- use UniProt to obtain annotation and functional information about a protein sequence
- use UniProt to identify and download homologues of a protein sequence
- generate a protein multiple sequence alignment using
MUSCLE - produce a Neighbour-Joining phylogenetic tree using
Simple Phylogeny - visualise a phylogenetic tree using
iToL
Please complete the workshop by answering the questions below in the formative quiz on MyPlace