2 Pairwise Sequence Alignment
Most sequence database search programs depend to some extent on pairwise sequence alignment - the process of identifying the mathematically optimal way to line up the symbols of two sequences in order so that they are as similar as possible.
Consider the two words COELACANTH and PELICAN. These words are obviously different, but they are also similar enough that we can shift their letters about to make an alignment between them. For instance, we might align the words as:
COELACANTH
-PELICAN--so that the E, L, C, A, and N letters are in the same columns. The only differences are then the matching of the O of COELACANTH with the P of PELICAN, and the need to insert gaps (- characters) to indicate where there is no corresponding letter, in the shorter word.
What if we were to align the words COELACANTH and PELICAN differently, like this?
COELACANTH
P-ELICAN--Is this a better or worse alignment of those two words than the one you originally saw? Why do you think this? What kind of rule would you devise to make a choice of which was the better alignment?

2.1 Pairwise Alignment is a Solved Problem
Pairwise sequence alignment is a solved mathematical problem. What that means is that, if we are given:
- two sequences
- a scoring scheme for matching symbols, such as:
- a match scores
+1(e.g.AandAare in the same column) - a mismatch scores
0(e.g.CandGare in the same column)
- a match scores
we can guarantee to find the alignment with the largest possible score, using an algorithm (i.e. a set of reproducible mathematical steps). The alignment with the largest score is considered the optimal alignment. Such algorithms find one of two kinds of optimal alignment: either a global or a local optimal alignment.
Given that we can find an alignment with a maximum score - the optimal scoring alignment - why should we believe that this is the best alignment that preserves biological function or information?
If we change the scoring scheme we might change the alignment, so what features of a scoring scheme might make it a better or worse representation of biological “reality”?
On this page you will carry out two pairwise alignments, one global and one local, of the same input sequences, to examine the differences between the two approaches. You will use the tools Needle and Water from the EMBOSS suite of sequence analysis tools (Madeira et al. (2022)).
2.2 Exercise 1: Global Pairwise Alignment
Global sequence alignment finds the optimal alignment across the full lengths of both sequences being aligned.
Given a scoring scheme, the Needleman-Wunsch algorithm is guaranteed to find an optimal global alignment between two sequences.
Using the two DNA sequences below, find their optimal global alignment using the EMBOSS Needle program at EMBL-EBI.
>sequence_1
ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA>sequence_2
TCACTTTAGAGGCTTGCAACCAGTACTTTAGAGGCTTGCTCAC2.2.1 Open the EMBOSS Needle webpage
Open the link https://www.ebi.ac.uk/jdispatcher/psa/emboss_needle in a web browser, to reach the landing page (Figure 2.8)
EMBOSS Needle landing page.
2.2.2 Enter the input sequences
Paste the two sequences given above into the two sequence input boxes, as shown in Figure 2.3.
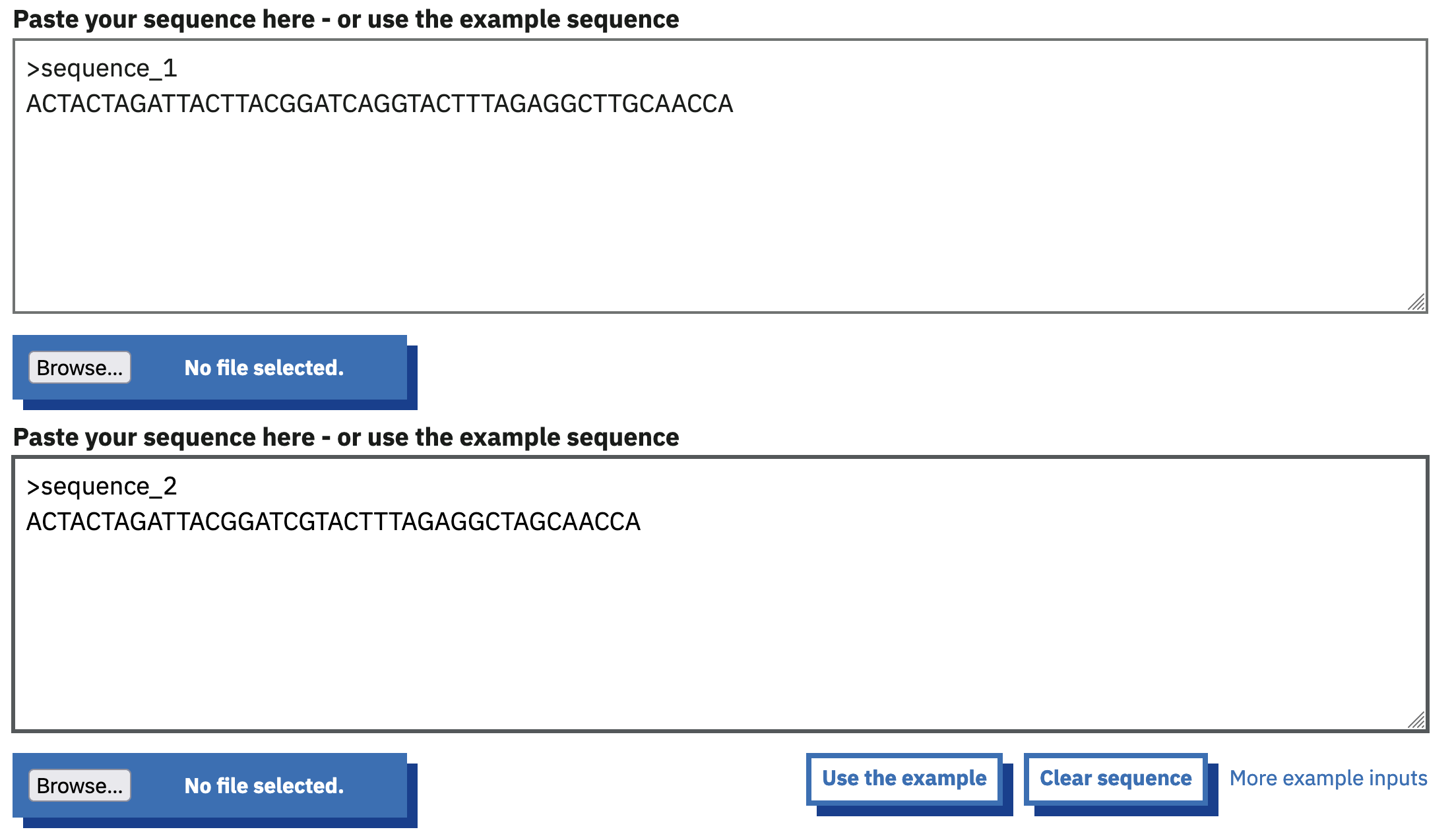
EMBOSS Needle page with two input sequences provided.
2.2.3 Set appropriate parameters
The two sequences you have entered are DNA sequences, so check the radio button marked DNA (Figure 2.4)

EMBOSS Needle input parameter set to DNA.
2.2.4 Run the alignment
Click on the Submit button at the bottom of the page to run the alignment. You can give your job a title, if you like (Figure 2.5).

EMBOSS Needle submit button.
A holding window will appear while the job runs (Figure 2.6), and after a short wait you will be presented with the results page (Figure 2.7).

EMBOSS Needle holding window.
EMBOSS Needle results window.
2.2.5 Interpret the results
The key result returned by Needle is, of course, the overall sequence alignment, which is provided at the foot of the results page:
sequence_1 1 ACTACTAGATTACTTACGGATCAGGTACTTTAGAGGCTTGCAACCA---- 46
|| ||||||||||||||||||||
sequence_2 1 --------------------TC----ACTTTAGAGGCTTGCAACCAGTAC 26
sequence_1 47 ----------------- 46
sequence_2 27 TTTAGAGGCTTGCTCAC 43- All bases from both sequences are represented in the alignment, even though the sequences are different lengths, and most bases are not found to align.
- Some bases in one sequence are aligned to the same base in the other - an identity - as indicated by the pipe symbol (
|) - Some bases in the upper sequence are not aligned to any bases in the lower sequence, and vice versa. Where this happens, the other sequence is padded out with gaps (
-). In those regions, the runs of bases may be referred to as insertions, and the missing sections represented as gaps may be called deletions.
2.3 Exercise 2: Local Pairwise Alignment
Local sequence alignment finds the optimal (i.e. highest scoring) alignment between two subregions of each input sequence.
Local alignment does not attempt to align all bases between the two sequences. Instead, it compares segments of all possible lengths between the two sequences to optimise the similarity score that it calculates. Only the best-matching regions are aligned.
Given a scoring scheme, the Smith-Waterman is guaranteed to find an optimal local alignment between two sequences.
Using the same two DNA sequences as before, find their optimal local alignment using the EMBOSS Water program at EMBL-EBI.
2.3.1 Open the EMBOSS Water webpage
Open the link https://www.ebi.ac.uk/jdispatcher/psa/emboss_water in a web browser, to reach the landing page (Figure 2.8)
EMBOSS Water landing page.
2.3.2 Enter the input sequences
Paste the two sequences you used for the Needle global alignment above into the two sequence input boxes, as shown in Figure 2.9.
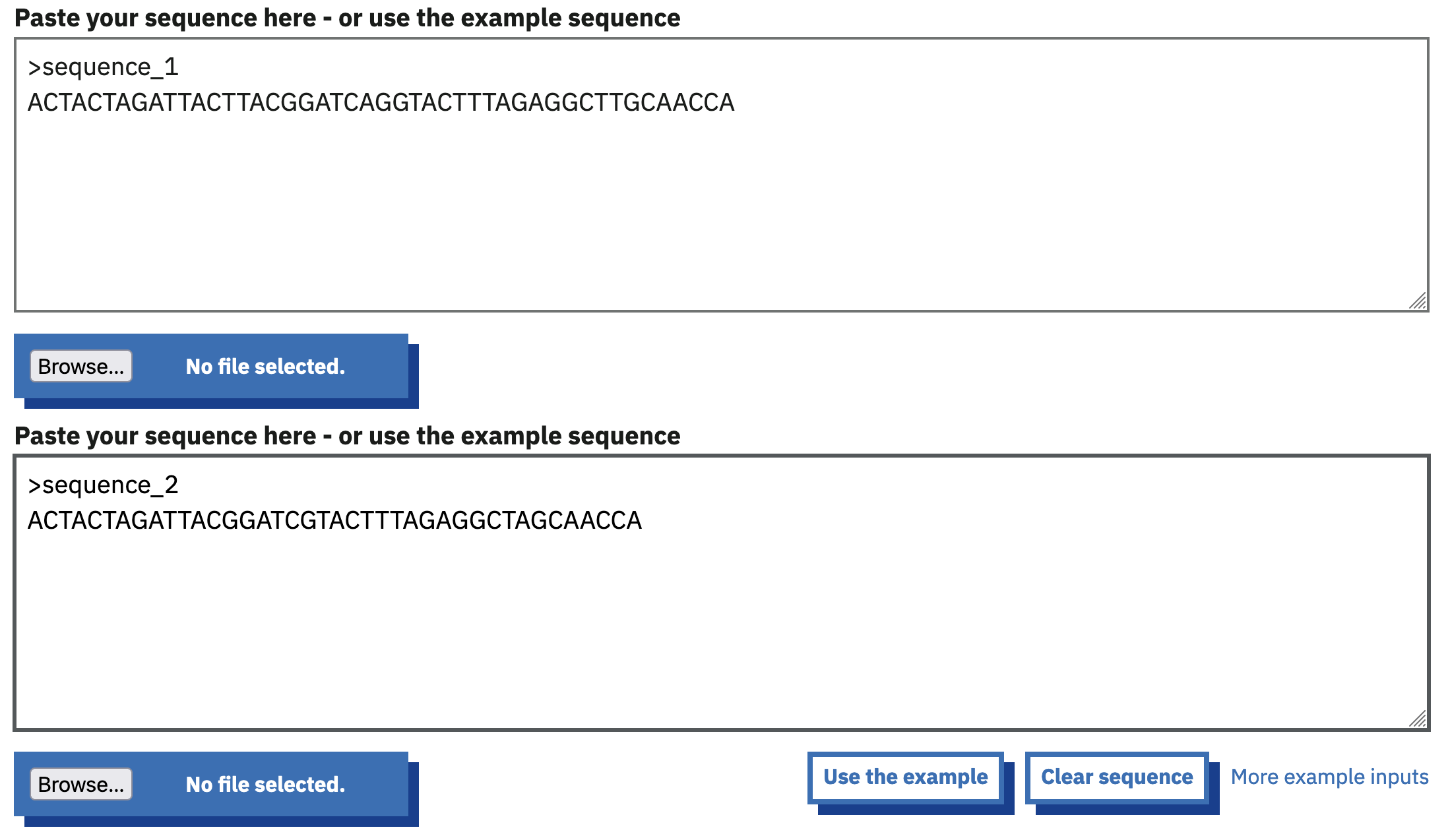
EMBOSS Water page with two input sequences provided.
2.3.3 Set appropriate parameters
The two sequences you have entered are DNA sequences, so check the radio button marked DNA (Figure 2.10)

EMBOSS Water input parameter set to DNA.
2.3.4 Run the alignment
Click on the Submit button at the bottom of the page to run the alignment. You can give your job a title, if you like (Figure 2.11).

EMBOSS Water submit button.
A holding window will appear while the job runs (Figure 2.12), and after a short wait you will be presented with the results page (Figure 2.13).

EMBOSS Water holding window.
EMBOSS Water results window.
2.3.5 Interpret the results (MyPlace Questions)
The key result returned by Water is again the overall sequence alignment, which once more is provided at the foot of the results page:
sequence_1 27 ACTTTAGAGGCTTGCAACCA 46
||||||||||||||||||||
sequence_2 3 ACTTTAGAGGCTTGCAACCA 22- Although the input sequences are the same ones that were used for global alignment, the local alignment is quite different: it is much shorter.
- Not all bases from either sequence are represented in the alignment - only bases
27to46ofsequence_1and bases3to22ofsequence_2. - Not all bases aligned in the global pairwise alignment are aligned in the local alignment.
Please answer the questions below in the formative quiz on MyPlace
Clicking on the green box should give you a hint to the answer, or where to find it.
Examine the second boxed section in the Needle output.
Examine the second boxed section in the Water output.
Consider the kinds of biological questions you might ask. When do you need to have the context of the full sequence information? When do you only need to know the best matching region? Which percentage identity result best represents the extent to which the two sequences actually resemble each other?
2.4 Summary
After completing this section you should:
- understand the difference between global and local pairwise alignments, and how to interpret their results
- know how to use the
EMBOSSWaterandNeedletools to obtain optimal pairwise alignments - be able to explain the output from the
WaterandNeedletools
Now you are ready to move on to using BLAST, which uses local sequence alignments to search large databases of reference sequences to find the best matches to an input query sequence.