1 Introduction
If two biological sequences are similar enough to each other, then we can hypothesise that they - or their products, in the case of coding sequences - may derive from a recent common ancestor, perform the same biological function, or adopt the same structural conformation.
1.1 Why do we care about biological sequence similarity?
It may be surprising, but we do not know what all genes in an organism do. In some organisms, we may not even know what most genes do, or even whether we know all the genes that are present.
Model organisms, such as Escherichia coli, are possibly the forms of life we understand best. But even the genome of E. coli O157:H7 - maybe the best-understood genome we know - contains ≈750 coding sequences (CDS) annotated as hypothetical protein, which means we have no evidence of what their functions are. This is 750/5067 ≈ 15% of the predicted proteome for that organism.
As biological sequences diverge through the actions of random mutation and nonrandom natural selection, they accumulate differences between them. The smaller the amount of time since two sequences diverged, the less time they have to accumulate these changes and, as a result, sequences from more recently-diverged organisms tend to be more similar to each other (we say they are more closely-related).
The fewer amino acid differences there are between two protein sequences, the less likely it is that the changes destroy or significantly modify its function. So, in general, the more similar two protein sequences are, the more likely they are to share the same functional activity, and to perform the same job for the organism they are found in. So, if we have a biological sequence of unknown function, and we can find a protein whose function we know whose sequence is similar enough to our sequence, then we can hypothesise that they have the same function.
Part of the reason the sequence of a protein enables its function is that the sequence dictates how the protein folds into its final shape. This shape places catalytic centres, and recognition surfaces for other proteins and small molecules, into position so that the biochemical function can happen. Similar protein sequences tend to fold into similar shapes, so proteins with similar sequences also tend to share similar structures.
Assigning a proposed function on the basis of sequence similarity to another sequence that has an annotated function is called functional annotation transfer. The vast majority of genes and gene products in public databases have functions assigned by this approach, but not by direct experimental verification. New sequences are added to these databases at a rate that far exceeds our ability to characterise their products experimentally, so this situation is likely to persist for some time.
The Critical Assessment of Functional Annotation (CAFA) is series of experiments and challenges designed to understand how well our computational methods of functional annotation match experimental reality (Zhou et al. (2019)).
1.2 Why does sequence similarity imply functional similarity?
If two proteins have the exact same sequence of amino acids, then they are essentially “the same thing.” We would expect them both to fold into the same structure, and to interact with the same molecules in the same ways. But the more changes we make to one of the sequences, the more likely it is that we will introduce a difference that results in a change to the structure or the functional activity of the molecule.
For example, the activity of an enzyme like chymotrypsin relies on the exact placement of three amino acid residues in a catalytic triad (Figure 1.1). If any one of these residues is substituted inappropriately, the enzyme no longer acts to catalyse the reaction that processes its substrate. However, several simultaneous substitutions can be tolerated elsewhere on the protein structure with no significant loss of catalytic function.
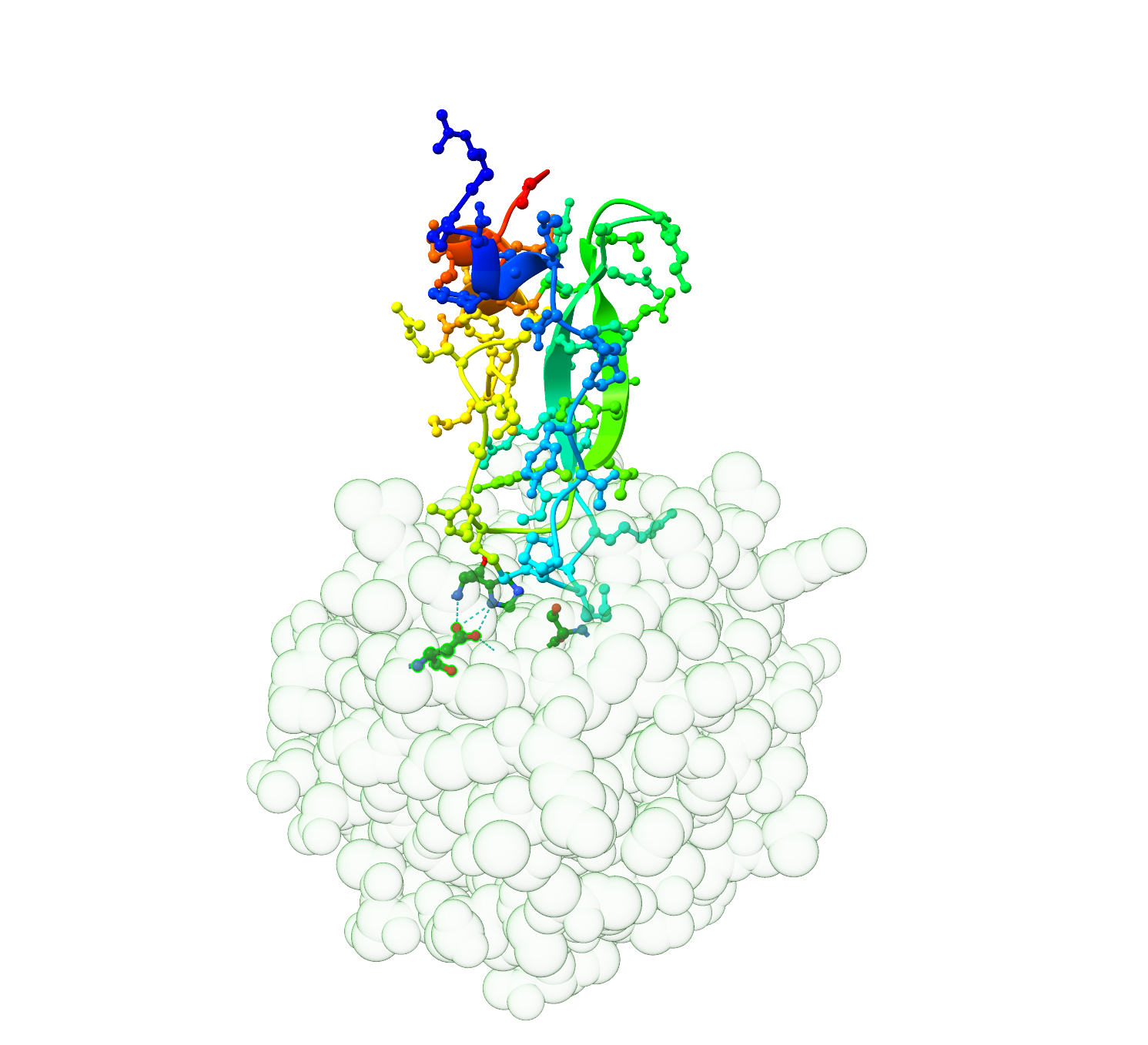
1.3 How similar is similar enough?
There is no single value of “similar” short of 100% exact identity that is guaranteed to be “similar enough” to imply identical function. What might qualify as “similar enough” depends on the scientific question you are asking, and will vary if you are asking questions like:
- Are these two biological sequences from the same organism?
- Do these two biological sequences share a single common ancestor?
- Do these two enzymes share the same catalytic function?
- Do these two enzymes catalyse the same reactions for the same substrates?
- Do these two proteins fold into the same \(\alpha\)-carbon backbone structure?
There are some heuristics (rules of thumb) that can guide you in how confident you should be in, say, assigning functional annotation or common structure on the basis of sequence identity. The table in Figure 1.2 (from Punta and Ofran (2008)) outlines some guidelines for interpreting functional similarity from sequence identity.
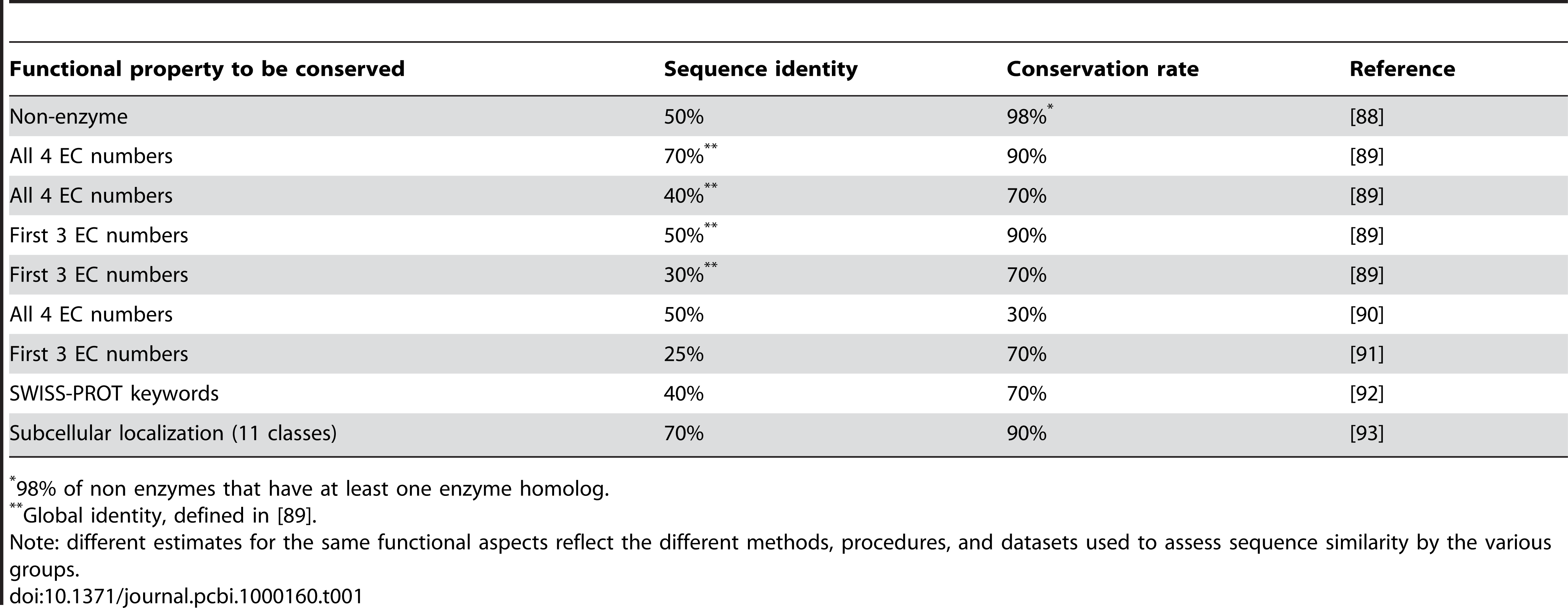
Similarly, we can often consider proteins sharing at least 40% amino acid identity to also have a similar \(\alpha\)-carbon backbone structure. Proteins sharing less than 20% identity are likely to have quite dissimilar structures, and the range 20-40% identity is considered a twilight zone where it is difficult to predict whether two proteins have a common structure (Figure 1.3).
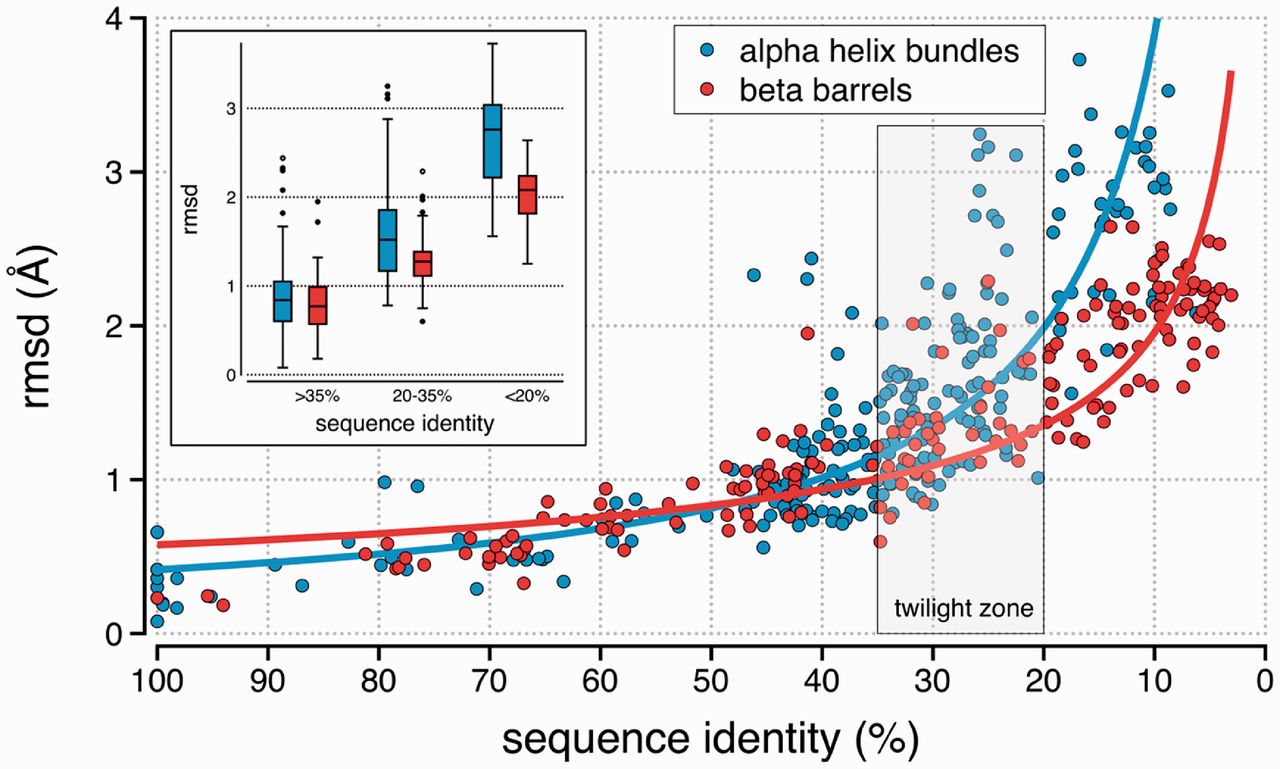
1.4 Where can we find similar sequences?
There are many publicly-available biological sequence reference databases available. Each has different contents, and may focus on a particular aspect of biology.
Always choose an appropriate sequence database suitable to answer your biological question.
1.4.1 INSDC
As a condition of publishing papers that describe new biological sequences - whether nucleotide or protein - those sequences must be deposited in a central public resource. For nucleotide sequence data, this is managed through the International Sequence Database Collaboration (INSDC), which is a collaboration between Japanese, US, and European organisations:
- DDBJ - the DNA Data Bank of Japan
- EMBL-EBI - the European Molecular Biology Laboratory - European Bioinformatics Institute
- NCBI - the National Center for Biotechnology Information, USA
These organisations share their data, so have approximately the same core sequence records (although accession IDs vary between them).
1.4.2 GenBank
The GenBank database contains an annotated collection of all publicly available DNA sequences, and is maintained by NCBI as part of the INSDC. The initial release of GenBank in December 1982 contained 606 nucleotide sequences. As of December 2023, GenBank contained 249,060,436 nucleotide sequences, and 2,863,228,552 additional sequences deriving from whole-genome sequencing. The number of bases in GenBank doubles approximately every 18 months.
1.4.3 RefSeq
NCBI also provides the RefSeq (NCBI Reference Sequence) database, which provides a comprehensive, integrated, non-redundant, consistently annotated set of reference nucleotide, transcript, and protein sequences.
1.4.4 UniProt
UniProt is a resource for protein sequence and annotation data. It comprises three databases:
- UniProt Knowledgebase (UniProtKB)
- UniProt Reference Clusters (UniRef)
- UniProt Archive (UniParc)
The main database of proteins is UniProtKB, and this is supported by UniRef - which clusters sequences on the basis of sequence identity - and UniParc, the archive of new, revised and obsolete sequences.
UniProtKB is itself comprised of two databases:
- Swiss-Prot, a manually annotated and reviewed set of sequences where every annotation has been checked by a human
- TrEMBL, the remaining set of known sequences, whose annotations were dervied by automated assignment, and have not been reviewed by a human
Manual annotation is time-consuming, and most proteins in UniProt have not been manually reviewed. As at 22nd February 2024:
- Swiss-Prot contains 570,830 manually-reviewed protein sequences
- TrEMBL contains 249,751,891 automatically annotated protein sequences
The current statistics can be found at UniProtKB
1.4.5 InterPro
Interpro is a service providing functional analysis of proteins by querying their sequences against a set of signature databases that contain patterns characteristic of particular functions or protein structures (Figure 1.4).
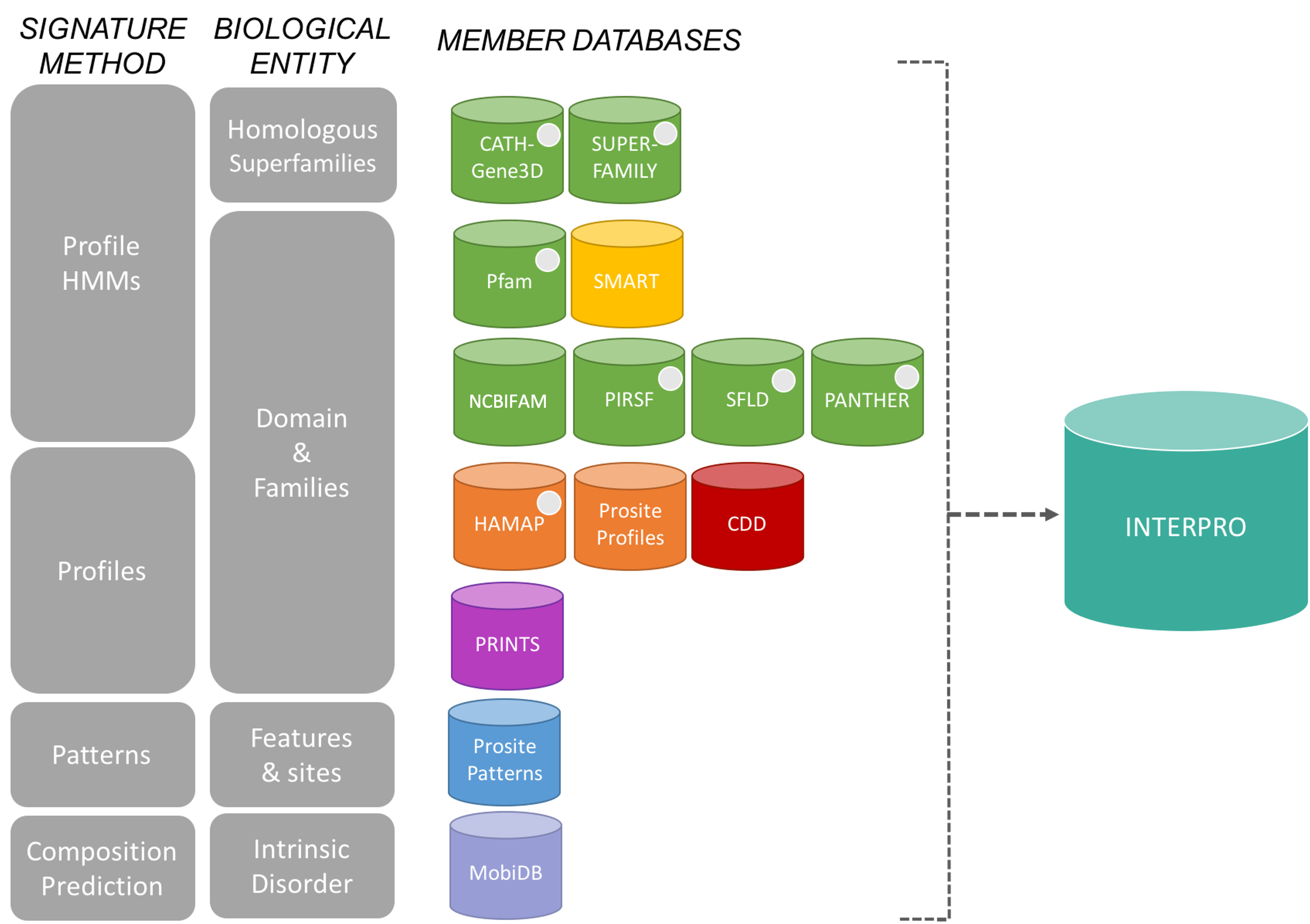
1.5 Summary
After reading through this section and following the links you should
- understand why it is important to be able identify similar sequences.
- be able to name some of the major sequence databases that help us use sequence similarity to understand the possible origins and functions of biological sequences and understand which might be most useful for a particular query.
Now you are ready to start using some of the bioinformatic tools for determining sequence similarity.