6 Scenario 3: Annotating a Metagenomic Contig
6.1 Introduction
You are working in a research group specialising in drug discovery. You have been investigating marine microbial communities using metagenomics in a search for sources of novel antibiotic candidates. You have carried out shotgun genome sequencing of an underwater microbial community and assembled a number of interesting contigs that likely contain novel genes. Now you need to annotate them. Today, you’re looking to annotate the sequence at this link.

Be sure to download the metagenome contig to your computer so that you can use it with NCBI BLAST to obtain a preliminary functional annotation.
To annotate your contig, you’re going to use NCBI BLAST’s database of model organisms to obtain a first draft annotation, to see if there might be some interesting functions present.
We would normally use specialised genome annotation software tools to annotate sequences like this, but we can illustrate the principles of genome annotation, and the advanced use of BLAST, with an exercise like this.
To annotate putative genes on this contig, you will BLAST its sequence against reference databases at NCBI. Carry out this search using the guide below (using the hints if you need them) and answer the questions in the formative quiz on MyPlace.
6.2 Analysis Steps
Follow the steps below to carry out the analysis for this scenario. If you need a reminder, please check with the example search in the BLAST introduction chapter. If you need more help, the green boxes below expand to give you a hint for what to do, and the orange boxes expand to give a more direct instruction.
6.2.2 Select the BLAST Tool
BLAST tool should I use?
Your query sequence is a contig - a nucleotide sequence - and you want to identify protein sequences that have functional annotations and are similar to regions on the contig, so this is a (translated) nucleotide vs protein search.
For a reminder, see Table 3.1
BLAST tool to use
This is a (translated) nucleotide vs protein search, so use the BLASTX tool.
6.2.3 Upload the query sequence
Click on the Browse… button.
Browse… button
Navigate to where you saved the metagenome contig file, and select it.
6.2.4 Set appropriate parameter choices
The NCBI BLAST webserver provides a database of sequences from model organisms.
The model organism database is small and non-redundant (no repeated sequences), and contains proteomes from a wide taxonomic range of organisms, making it ideal for preliminary annotation. The small size of this dataset means that results are returned quickly and concisely.
The NCBI BLAST webserver allows you to query against a small non-redundant database derived from model organisms (the landmark database). You should use this for preliminary annotation with BLASTX.
Select the Model Organisms (landmark) database
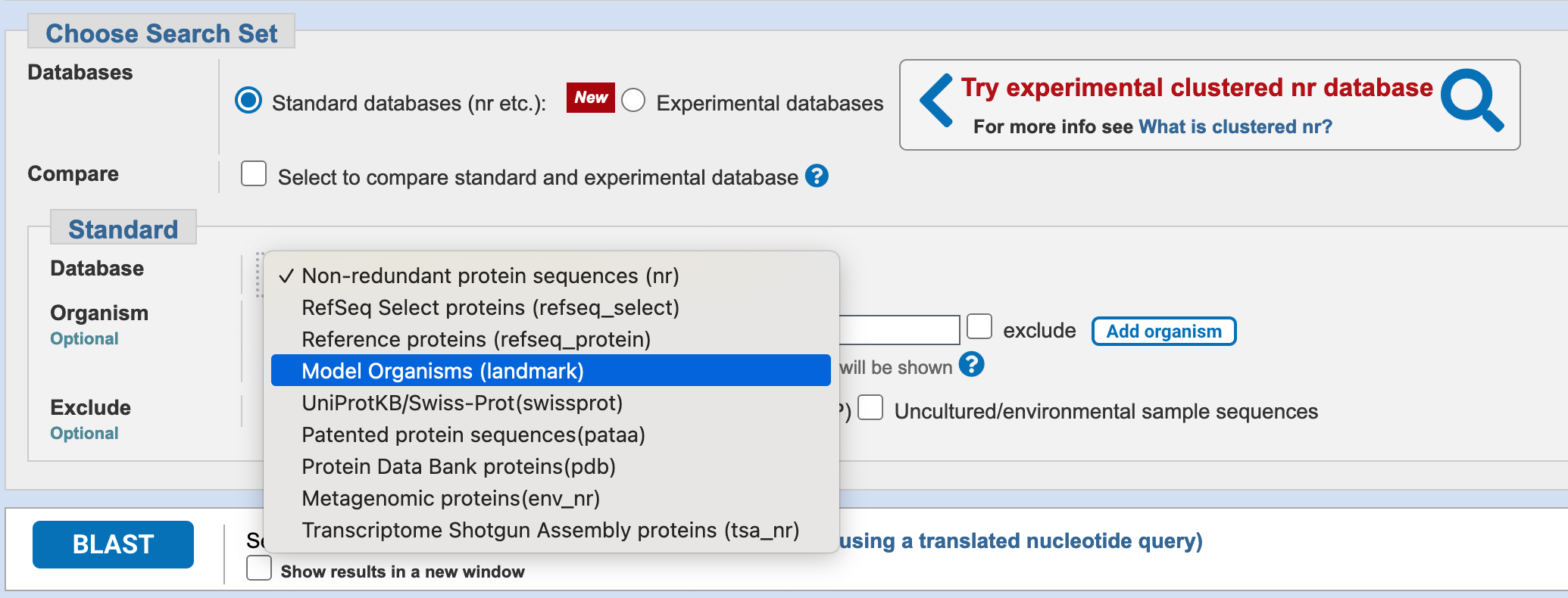
Model Organisms (landmark) database
6.2.5 Run the BLAST search
BLAST search
Click on the BLAST button.

BLAST button
6.2.6 Interpret the BLAST report (MyPlace Questions)
Your metagenomic contig likely contains multiple genic or protein-coding regions. The matches to these will all be present in the report at the same time, so you will need to approach the interpretation of this report differently from those in scenarios 1 and 2. You may find it useful to start your interpretation by looking at the Graphic Summary tab.
Please answer the questions below in the formative quiz on MyPlace
Clicking on the green box should give you a hint to the answer, or where to find it.
Check the Graphic Summary tab, and count the number of distinct regions of your query with matches in the database.
Check the Graphic Summary tab, and count the number of distinct regions of your query with matches in the database, and the appropriate colour of annotation in the graphic.
Check the Descriptions tab and identify the top hit.
Check the Graphic Summary tab, and click on the topmost alignment at the appropriate region. Click on the Alignment link in the box that appears.
Check the Graphic Summary tab, and click on the topmost alignment at the appropriate region. Click on the Alignment link in the box that appears.
Check the Graphic Summary tab, and click on the topmost alignment at the appropriate region. Click on the Alignment link in the box that appears.
Check the Taxonomy tab, and make a judgement based on the Score and the number of hits in the report.
6.3 Stretch Activities
These activities are not necessary for the assessment, but use more features of the NCBI services to provide more information relevant to interpreting your BLAST results. Open the orange boxes to see the questions, and use the green boxes to find some answers.
6.3.1 Refine your search
From your analysis above you will have identified a candidate taxonomic assignment for your organism. With this, you can repeat your query against the much more comprehensive nr database, but refining the search by filtering so that it only reports sequences from the corresponding taxonomic Kingdom. You can also refine your search against the pdb database of solved protein structures, to investigate the molecular function of your annotations by predicting conserved domains and visualising representative structures.
nr
Return to your BLAST query by clicking on the Edit Search button at the top of the report.
BLAST report Edit Search button
Select the nr database and restrict the taxon by entering the appropriate value into the Organism field.
BLASTX search
Click on the BLAST button.

BLAST button
- What taxon did you enter in the
Organismfield? - Do you find the same number of potential protein-coding regions as in the initial search?
- What is the percentage identity of the highest-scoring match?
- Are the annotations of the protein matches similar to the previous search?
- What is the most likely taxonomic classification for the query organism?
- What kinds of differences between the databases used for the searches have affected your results, and how has the quality of the results differed?
nr
- Archaea (taxid:2157)
- No - there are fewer: only three (3)
- 99.89%
- Some are, some are not. There is one match to the region spanning base 2500 in the query, not annotated the same way as the previous search. The regions spanning bases 7500, and 10000 have similar functional annotations.
- The precise taxonomy at genus or species level is not clear from this search. Most matches are to sequences only annotated at Phylum level (Euryarchaeota) or that have not been formally assigned taxa (Candidatus).
- There are several points to consider:
- The
Model Organisms (landmark)database is small (it contains only 27 genomes), so is not very comprehensive, even though it contains representatives across the entire tree of life. We should not expect very close sequence matches as a result, but the search should be quick. - The
nrdatabase, even filtered to provide results only for archaea, contains many more genomes and is much larger. As we have “tuned” the database to organisms similar to the query, we should expect many close matches at the expense of a slower search. - The
Model Organisms (landmark)database contains well-annotated genomes and we should expect clear indications of molecular function for the matches. - The
nrdatabase contains all relevant sequences in the NCBI database, many of which have not been annotated or may have been annotated with less certainty. The annotated functions of matches here may be less clear. - The
Model Organisms (landmark)database contains relatively few organisms so we should not expect an accurate prediction of taxonomy for our query. - The
nrdatabase contains all relevant sequences in the NCBI database and is likely to place our query organism more accurately alongside close relatives, even if the taxonomy of those relatives is uncertain.
pdb
Return to your BLAST query by clicking on the Edit Search button at the top of the report.
BLAST report Edit Search button
Select the pdb database and do not restrict the organism range.
pdb database
Click on Algorithm parameters to reveal more options and modify the Expect threshold to read 1e-6 to include only strong matches.
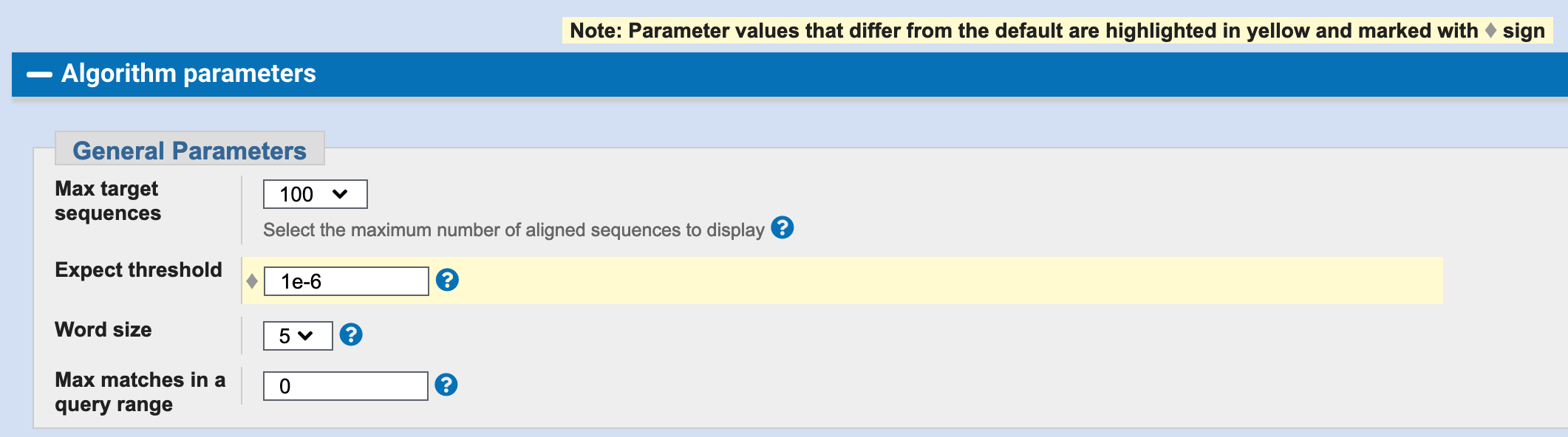
Expect threshold
Click on the BLAST button.

BLAST button
- What is the molecular function of the top hit?
- What organism does the top hit come from?
- What is the percentage identity of the top hit?
- What is the PDB accession number of the top hit?
Click on the accession number for the top hit to open the protein record for that page in a new tab. Click on the Identify Conserved Domains link under Analyse this sequence on the right-hand side of the page. This will take you to a prediction of the conserved domains

Analyse this sequence panel
- What is the predicted domain superfamily?
Click on the link at the top of the prediction page (“Conserved domains on [gi|657231874|pdb|3WQY|A]”) to return to the protein description page. Then click on the thumbnail image under Protein 3D Structure on the right hand side of the page to open the protein structure page. Click on the View in iCn3D link for the top match.
- How many protein structures were available?
- What is the RCSB-PDB accession for the structure you are viewing?
- How many copies of the enzyme are visible in the structure?
- Are any other molecules visible in the structure? If so, what are they?
pdb
- Alanine-tRNA ligase
- Archaeoglobus fulgidus
- 42.74%
- 3WQY_A
- A-tRNA_syn_arch superfamily
- Four (4)
- 3WQZ
- Two copies of the enzyme are visible, as chains A and B.
- Yes. There is a 75bp RNA molecule (chain C), and two molecules of A5A (‘5’-O-(N-(L-Alanyl)-Sulfamoyl)adenosine). There are also two zinc ions.
6.4 Summary
After successfully working through this scenario, you should be able to
- use
BLASTto obtain preliminary identifications for a section of genome - identify the likely taxonomic origin of a genomic sequence using
BLAST
If you completed the stretch activities you should be able to
- modify your search parameters for the same query, choosing the most appropriate database with or without taxon filters to answer a specific biological question
- explain how the choice of database affects the accuracy and comprehensiveness of your results
- use
BLASTresults at NCBI to obtain domain and structural information relevant to your protein - use the NCBI
BLASTservice to help visualise structures of homologues to your protein