6 Inspect Read Quality
Using good quality input data improves our chance of getting a good quality output assembly.
We call the set of sequenced reads we obtain from the sequencer the raw reads. Modern sequencing technologies usually give us high quality data, but it is still possible for some low-quality read sequences to be produced among the raw reads. So that these do not detrimentally affect the quality of our assembly, we first assess the quality of our read data, and then remove low-quality reads and low-quality parts of read sequences. The resulting dataset is often called the cleaned reads or processed reads.
It is good practice always to inspect the quality of your sequencing read data, and remove low-quality reads, before assembly.
When describing your sequencing experiment in a manuscript or dissertation, you should always state:
- The sequencing technology used, and the sequencing platform
- The number of raw reads obtained, and a measure of average read length
The main kind of low-quality data we might have in our raw reads are:
- Contamination: reads that derive from an organism we didn’t intend to sequence; these are usually identified by querying against a database and removed
- Low-confidence basecalls: reads containing base calls that the basecaller is unsure about; these are usually found towards the end of the read sequence and are removed by trimming the read
- Low-confidence reads: reads where a large proportion of the base calls are low quality; these are usually removed
- Adapter sequence: these are sections of sequence left over from library preparation and do not derive from the sequenced organism; they are removed by trimming
This part of the workshop will cover the use of FastQC to inspect the quality of your sequence reads.
6.1 Using FastQC
- Navigate to the
FastQCtool using theToolssidebar inGalaxy
- You can use the
search toolsfield to findFastQC - Alternatively, you will find it under
GENOMIC FILE MANIPULATION/FASTA/FASTQin the sidebar
- Select the
FastQCtool - Run the
FASTQCtool on each of yourFASTQinput files
- These are the
trimmed_pe_aln.qsorted.mapped.fixed.1.fastq.gzandtrimmed_pe_aln.qsorted.mapped.fixed.2.fastq.gzfiles. - With the
FastQCtool options in the mainWorkspacewindow, select the file you want to runFastQCon. - Click on the
Run Toolbutton.
Make sure you run FastQC on both the forward and reverse read sets.
FastQC to assess the quality of your sequencing reads
FastQC takes a few moments to run.
When the run is complete, click on the eye icon of the Webpage result to inspect the FastQC output.
You can press and hold CTRL (on a PC) or CMD ⌘ (macOS) when clicking the eye icon to open the result in its own window.
6.2 FastQC output
The output of FastQC tells us a lot about the quality of our sequencing data, and we would typically use it to identify problems with our sequencing data. Some important sections are described below, and you can read more about how to diagnose quality issues in your dataset at the links below:
FastQC output in Galaxy
6.2.1 FastQC Summary
The FastQC summary (Figure 6.1) provides an overview that indicates where there may be areas of concern in your data. Each section of the report receives a flag indicating either that it gets a pass (green, with a tick), a warning (amber, with exclamation mark), or a fail (red, with a cross) mark.

FastQC summary showing passes for most sections of the report including “Basic Statistics,” warnings for some sections including “Overrepresented sequences,” and a fail for “Sequence Duplication Levels”
6.2.2 Per base sequence quality
The per base sequence quality plot (Figure 6.2) presents the ranges of individual base quality calls across the lengths of the input sequence reads. Higher scores (near 40) indicate better quality.
For good-quality sequence read data, we are looking for a graph where the mean quality (see Figure 6.2) is always in the green area, with a score greater than 29.
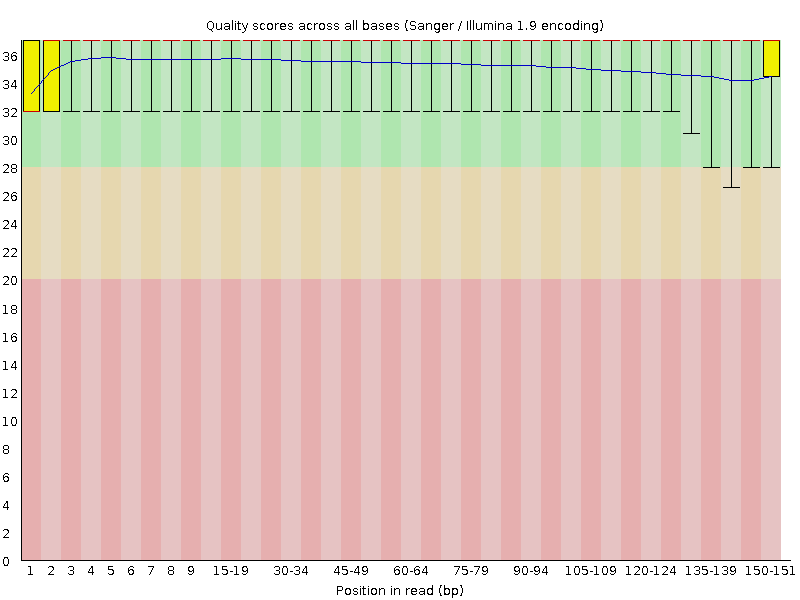
FastQC indicating high quality sequencing reads. Quality score ranges are presented as a boxplot, with the mean drawn as a line connecting each boxplot.
It is not unusual for the per-base quality scores to fall quite steeply towards the end of the reads, with Illumina and other sequencing technologies.
It is also typical for the quality score to be slightly lower in the first 5-7 bases of the read.
If mean quality per base falls below 29, there may be a problem with the sequencing data.
6.2.3 Per sequence quality scores
The per sequence quality score (Figure 6.3) summarises the distribution of high quality (higher scores, near 40) and lower quality reads in the complete dataset.
In good quality sequence data, we tend to see a sharp peak towards the right hand side of the plot.
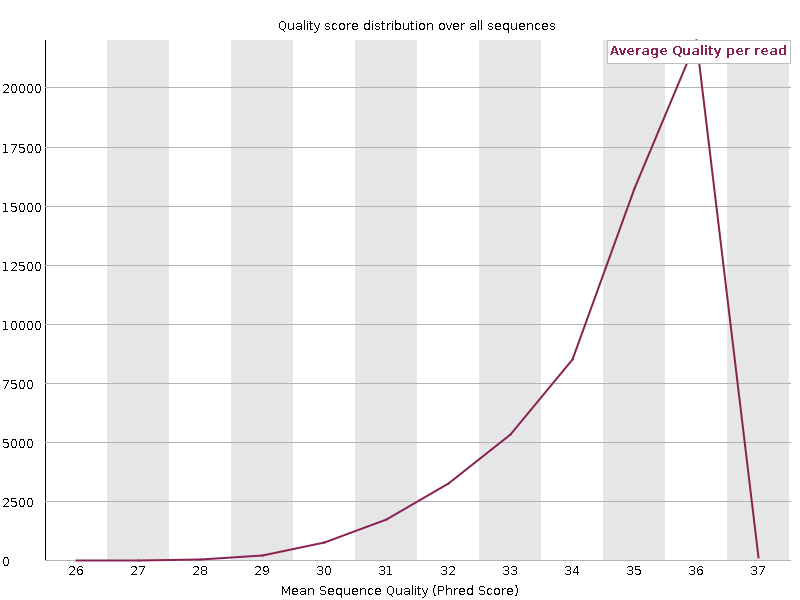
FastQC indicating overall high quality sequencing reads.
If the per sequence quality score appears flat, or plateaus towards the left hand side of the plot, there may be a problem with the sequencing data.
- The
FastQCSequence Duplication Levels section is marked as a fail in one of the output result files. Why do you think this is?
- Read the description of the Sequence Duplication Levels result section in the MSU Tutorial.
- Read the description of the provenance of your read data in Chapter 1
Your sequence reads were obtained using RNA sequencing, and very deeply sequenced the virus genome. The high abundance of virus sequence reads (and the low abundance of any reads that do not derive from the virus) looks unusual compared to the results of a standard DNA genome sequencing experiment, so is marked as “fail” by FastQC.
6.3 What do we do about sequencing data problems?
FastQC does a great job of alerting us to problems with our sequencing data sets. But, by itself, it cannot remedy these problems.
Modern sequencing methods produce so much read data that the usual approach to excluding poor data is either to exclude (or drop) the read itself, or to trim (throw away) the section of the read sequence that is low quality.
A popular tool for removing poor quality read data is trimmomatic and you will meet this in Chapter 7.