7 Clean Raw Read Data
As you have seen in Chapter 6 sequence read data can be of variable quality. The usual approach to improving the overall quality of a dataset is to discard low quality reads, and low-quality sections of reads. This is referred to as cleaning raw read data.
Trimmomatic is a commonly-used software tool for cleaning raw read data that
- identifies and excludes (drops) low-quality reads
- trims (deletes) any remaining Illumina adapter sequences
- trims (deletes) any low-quality read ends
It is good practice always to use a tool like trimmomatic to clean your raw sequencing data.
When reporting how you cleaned your sequence data in a manuscript or dissertation, you should always state:
- The software tool you used, with its version number and a citation of the paper describing it (if available; provide a URL to the software if there is no paper)
- The parameters used when running the tool (if default parameters were used, state this)
- The number of reads that remained after cleaning
Tools like trimmomatic return two kinds of cleaned read data:
- R1-paired, R2-paired: these are high quality paired-end reads
- R1-unpaired, R2-unpaired: these are single reads, where one member of the read pair was low quality
This part of the workshop will guide you through using trimmomatic to clear your sequence read data.
7.1 Using trimmomatic
- Navigate to the
trimmomatictool using theToolssidebar inGalaxy
- You can use the
search toolsfield to findtrimmomatic, or look under theFASTA/FASTQsection in the sidebar
- Select the
trimmomatictool - Make sure that
Paired-end (two separate input files)is chosen - Select the forward and reverse raw read sets (
trimmed_pe_aln.qsorted.mapped.fixed.1.fastq.gztrimmed_pe_aln.qsorted.mapped.fixed.2.fastq.gz) as theInput FASTQ files - Set
Perform initial ILLUMINACLIPtoYes; this will trim any remaining Illumina adapters from the read sequences - Under
Adapter sequences to useselectTruSeq3 (paired-ended for MiSeq and HiSeq) - Click
Run Tool
trimmomatic to clean raw sequencing reads
7.2 trimmomatic Output
Cleaning your data with trimmomatic generates four output files (Figure 7.1).
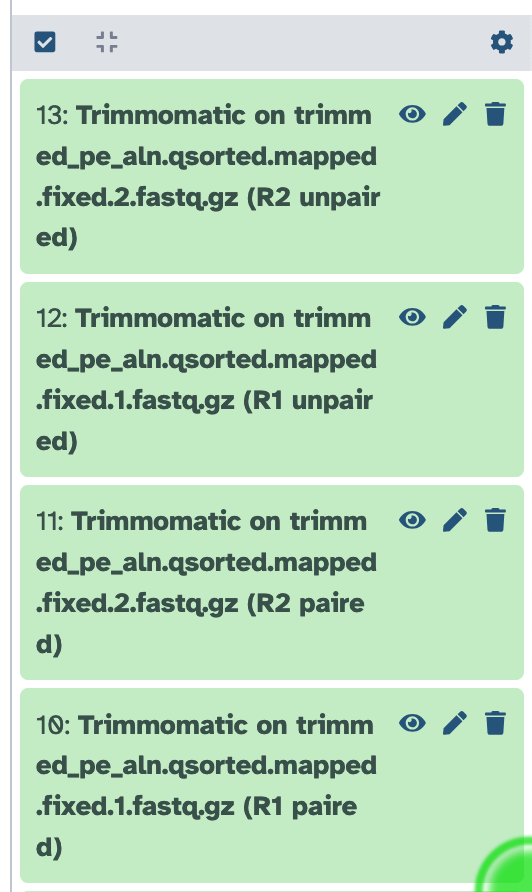
FastQC output files in Galaxy. From the top: unpaired reverse (R2) reads, unpaired forward (R1) reads, paired reverse (R2) reads, and paired forward (R1) reads.
You can inspect the contents of these files by clicking on the filename in the History sidebar, and also by clicking on the corresponding eye icon to view the trimmed reads in the workspace.
trimmomatic output files
7.3 Next steps
Now that we have clean, high-quality sequencing reads, we can move on to assembling the SARS-CoV-2 genome, in Chapter 8.