1 Introduction
1.1 Workshop Scope
Today you will be:
- assembling, annotating, and visualising the first SARS-CoV-2 genome to be sequenced (isolated in Wuhan, China, in January 2020).
- comparing the first SARS-CoV-2 genome to an earlier coronavirus (SARS 2003)
- comparing the first SARS-CoV-2 genome with a SARS-CoV-2 genome that was sequenced later in the pandemic
- interpreting the biological signifance of differences between the genomes, particularly in the region of the spike (S) protein.
1.2 COVID-19
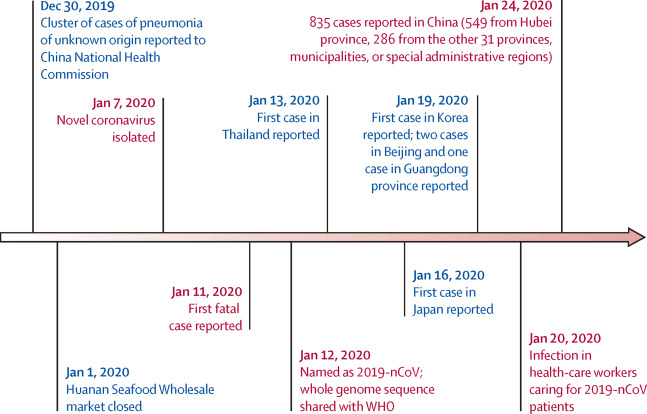
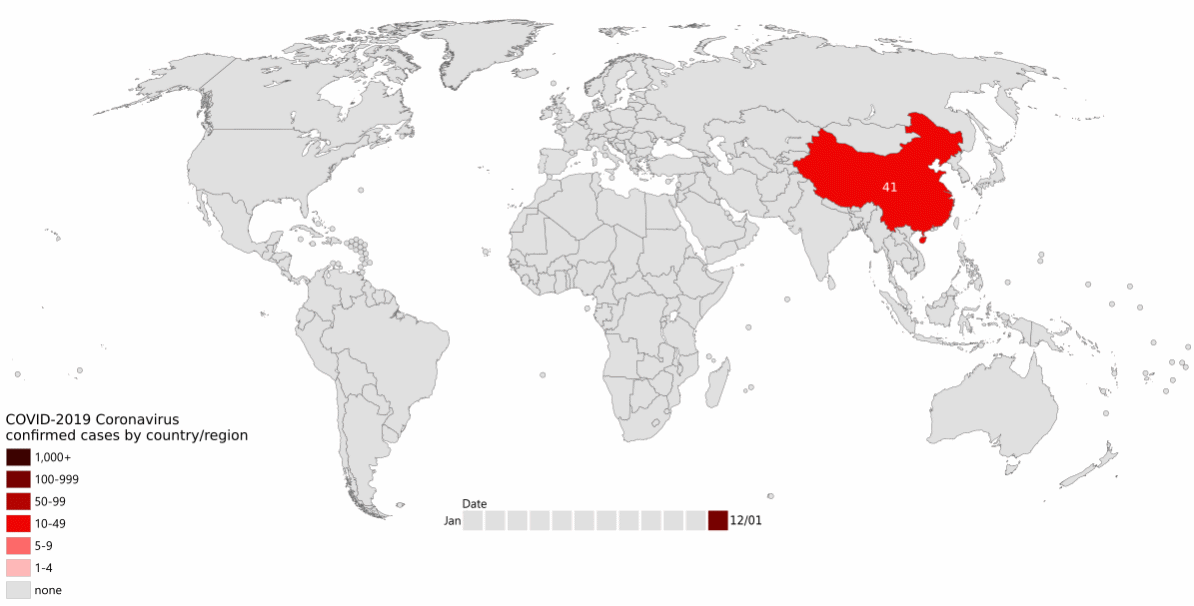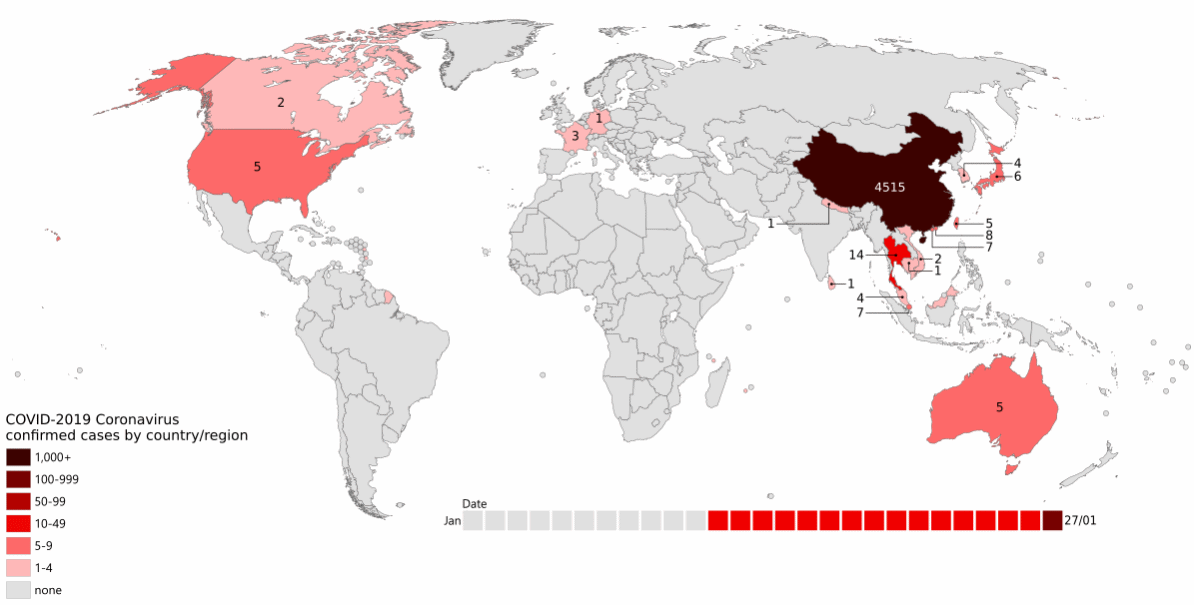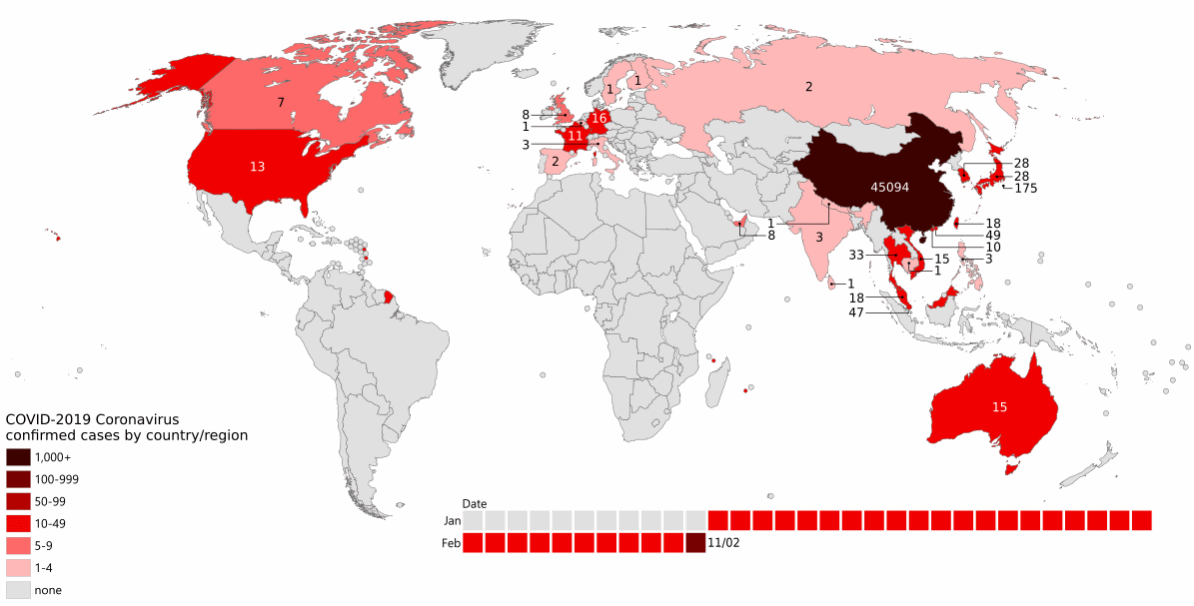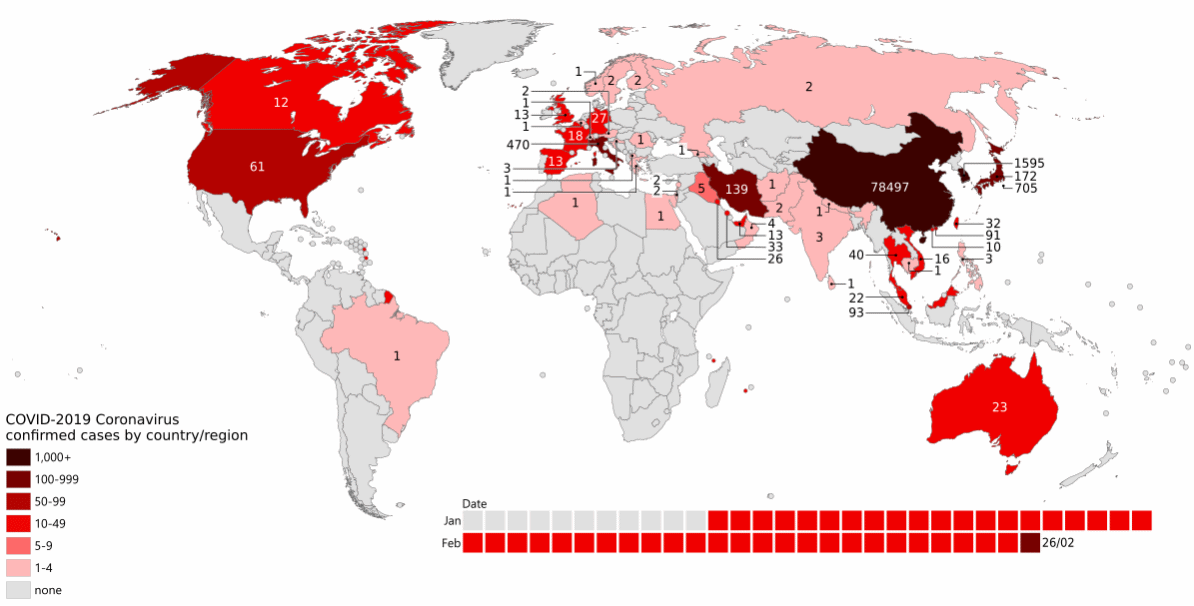
The first cases of a pneumonia of unknown causes began to emerge in Wuhan, China, in December 2019. The novel coronavirus SARS-CoV-2 was isolated by early January, and by the end of January 2020 human-to-human transmission had been confirmed, and cases had been reported in Thailand, Japan, the USA, and several other countries (Figure 1.1, Wang et al. (2020)).
By the end of February, cases had been identified across the Northern hemisphere, and a new hotspot was emerging in Italy (Figure 13.1).
By October 2021, it was estimated that COVID-19 had caused over 5mn deaths worldwide, as well as severe social and economic disruption, including the largest global recession in almost a century.
Despite this huge impact, societal and scientific efforts including lockdowns, mathematical modelling, and novel vaccine development - much of it supported by widespread genome analysis of the kind you will perform today - helped avoid even worse outcomes. COVID-19 vaccines are estimated to have prevented an additional 14-20mn deaths worldwide in 2021 (Watson et al. (2022)).
In May 2023, the World Health Organisation (WHO) ended their Public Health Emergency of International Concern (PHEIC), but the SARS-CoV-2 virus remains in circulation. As we begin this workshop, SARS-CoV-2 has claimed over 7mn lives (Mathieu et al. (2020)).
1.3 SARS-CoV-2 Spike (S) Protein
The spike (S) protein of SARS-CoV-2 plays a key role in host cell receptor recognition and cell membrane fusion (Huang et al. (2020)). The protein comprises two subunits:
- S1: a receptor-binding domain that recognises and binds ACE2 (angiotensin-converting enzyme 2)
- S2: a domain that mediates membrane fusion between the virus and the cell by forming a structural bundle
These interact with the host cell as illustrated in Figure 1.3.
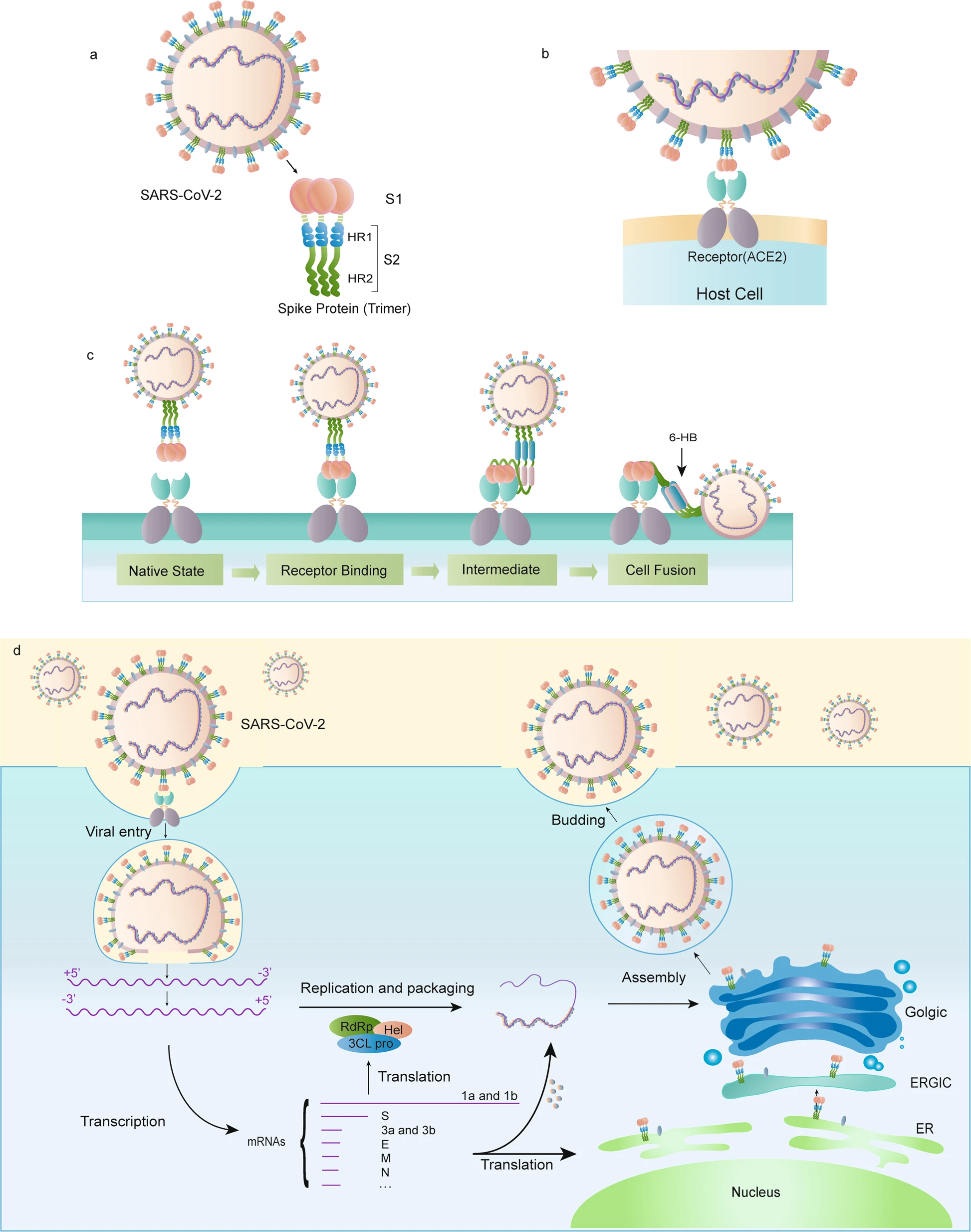
When you assemble, annotate, and visualise the SARS-CoV-2 genome in this workshop you will find the spike protein annotated as in Figure 10.1.

The spike protein is not the only important surface protein for SARS-CoV-2. The envelope (E), membrane (M) and nucleocapsid proteins are also essential for the virus to interact with the host, as shown in Figure 1.5, and you will identify these in your analysis today, too.
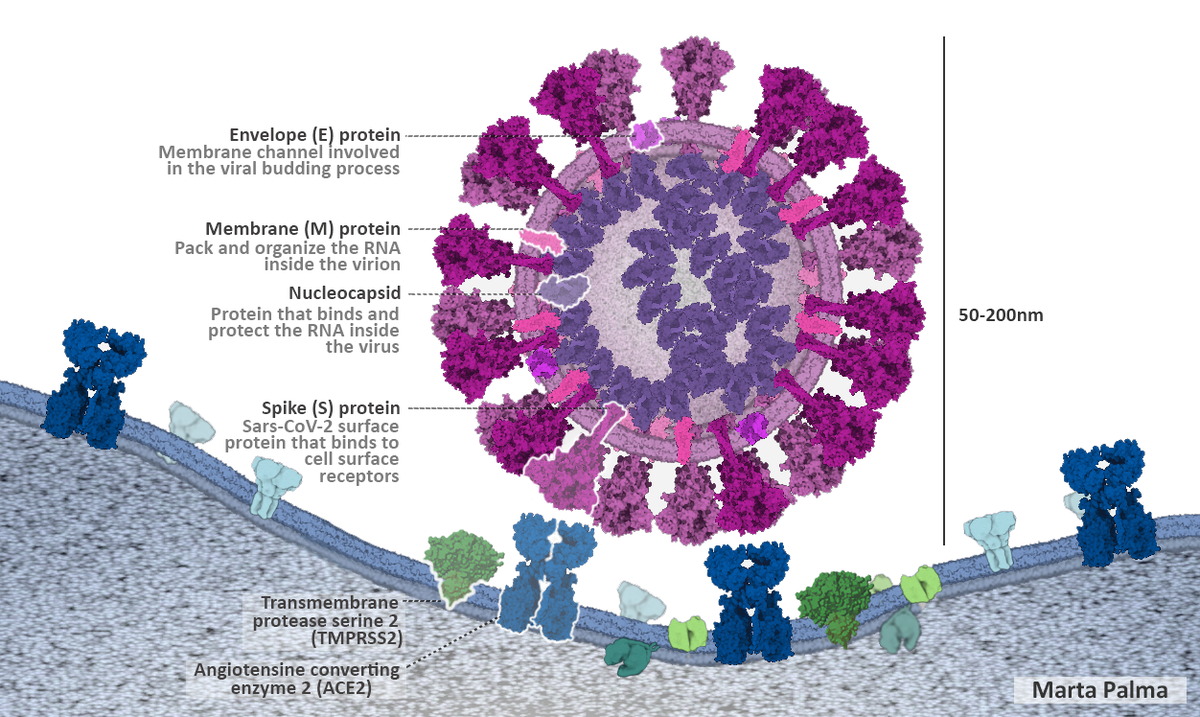
You can find an interactive collection of publicly-available SARS-CoV-2 protein structures at RCSB/PDB, the public repository for biological structural data.
1.4 Data Provenance
Modern scientific research, and this workshop, are both made possible due to the dedication of scientists around the globe to FAIR (Findable, Accessible, Interoperable, and Reproducible) principles for scientific data management.
Throughout the COVID-19 pandemic, SARS-CoV-2 sequence and structural data was released through public repositories including NCBI, GISAID, and RCSB/PDB. The data shared through these resources enabled scientists across the world to conduct research that reduced suffering and hastened the end of the pandemic. These practices were established before COVID-19, and continue to support and empower scientific research globally.
1.4.1 The first sequenced human COVID-19 case
The first SARS-CoV-2 genome was obtained from a sample of human lung in January 2020. The sample was sequenced and submitted to NCBI as SAMN13922059. It is recorded as “human lung metagenome” because the entire material - human, virus, and everything else - was sequenced as a single sample. The work as described in Wu et al. (2020) is outlined below.
- The lung was lavaged (washed/rinsed) and the fluid collected
- Total RNA was extracted from the fluid, and an RNA sequencing library constructed (including depletion of ribosomal RNA)
- The library was sequenced on an Illumina MiniSeq machine, to give 150bp paired-end reads
- The resulting reads were cleaned and assembled using Megahit (Li et al. (2015)) and Trinity
- The longest assembled contig was identified as a coronavirus, and used to design primers for PCR-RACE (Rapid Amplification of cDNA Ends, Pal (2022), Figure 1.6) which allowed researchers to sequence the ends of the virus genome
- The cleaned reads were remapped against the full virus sequence
- Why did the researchers extract RNA and not DNA?
- Why do you think the researchers depleted the ribosomal RNA?
- SARS-CoV-2 is an RNA virus, so we must sequence RNA in order to recover the virus genome. Sequencing DNA would not include any virus sequence.
- There is so much ribosomal RNA in biological material that, if it was left in the sample, it would dominate over the viral material and any human transcripts. Most of the resulting sequence would describe the ribosome, not the virus or host genome, making the virus more difficult to detect. Depleting ribosomal RNA enhances sensitivity for detecting viral RNA.
1.4.2 SARS-CoV-2 read data for this workshop
For this workshop, we downloaded the 57mn raw sequencing reads from Wu et al. (2020), which are available at NCBI under accession SRR10971381, and filtered out all reads that matched the human genome. The remaining reads in the files below are those you will use to assemble the SARS-CoV-2 genome.
1.4.3 SARS-CoV 2003 genome data for this workshop
The first comparator genome in the workshop is that of SARS coronavirus Urbani. This coronavirus emerged in Guangdong, China, in late 2002 and was the subject of a World Health Organisation global alert, which named it SARS (Severe Acute Respiratory Syndrome). This strain of SARS-CoV was sequenced in 2003 (Rota et al. (2003)) and has accession AY278741.1 at GenBank/NCBI.
In the workshop, you will use the following files:
1.4.4 March 2020 Dutch SARS-CoV-2 isolate
The second comparator genome you will use in this workshop is a SARS-CoV-2 variant sequenced in the Netherlands in March 2020, four months after the first known human cases were identified. The sample has accession SAMEA6847958 and was obtained through active surveillance during the COVID-19 pandemic (Oude Munnink et al. (2020)).
The data describes a set of Oxford Nanopore (ONT) GridION reads, which are available under accession ERR4164769 at SRA/NCBI. In this workshop, you will use the following file: